Information Links
Related Conferences
Previous Issues Volume 9, Issue 1 - 2025
The Role of the Antioxidant Barrier in the Development of Arrhythmia in Structural Changes in the Heart
Khabchabov Rustam Gazimagomedovich1,*, Elmira Rashitbekovna Makhmudova2
1Associate Professor of the Department of Cardiology, Emergency Care and General Medical Practice, Federal State Budgetary Educational Institution of Higher Education “Dagestan State Medical University” of the Russian Ministry of Health, Lenin Square 1, Makhachkala 367000, Russian Federation
2Assistant of the Therapy Department, Federal State Budgetary Educational Institution of Higher Education “Dagestan State Medical University” of the Russian Ministry of Health, Lenin Square 1, Makhachkala 367000, Russian Federation
*Corresponding Author: Dr. Khabchabov Rustam Gazimagomedovich, PhD, Associate Professor of the Department of Cardiology, Emergency Care and General Medical Practice, Federal State Budgetary Educational Institution of Higher Education “Dagestan State Medical University” of the Russian Ministry of Health, Lenin Square 1, Makhachkala 367000, Russian Federation, Tel: +7 (963) 413-67-20, ORCID: 0000-0002-8174-3900, E-mail: [email protected]
Received Date: June 20, 2025
Published Date: June 30, 2025
Citation: Khabchabov RG, et al. (2025). The Role of the “Antioxidant Barrier” in the Development of Arrhythmia in Structural Changes in the Heart. Mathews J Cardiol. 9(1):40.
Copyrights: Khabchabov RG, et al. © (2025).
ABSTRACT
The mechanism of development of paroxysmal tachycardia, flutter and fibrillation of the heart with structural changes (remodeling) of the myocardium remains unknown to this day! We know that many of you will be indignant and say “everything is already known”! We want to introduce you to new mechanisms of these cardiac arrhythmias, which are closely related to each other. In addition, you will learn: what is an “antioxidant barrier”; how a large wave of macro re-entry is formed during flutter; you will understand how flutter differs from paroxysmal tachycardia.
Keywords: Antioxidant Barrier, Оxidative Stress, Paroxysmal Tachycardia, Flutter, Fibrillation of the Heart.
INTRODUCTION
Cardiac fibrillation is the most dangerous type of arrhythmia; if fibrillation occurs in the ventricles, then the person has already stepped one foot into the world of the dead. And if fibrillation occurs in the atria of the heart, then serious complications may arise, such as the formation of blood clots in the heart cavity, which are often carried by the blood flow to other organs, primarily the brain, causing blockage of the lumen and cessation of blood flow through the vessel, which leads to the development of ischemia, up to a stroke. In addition, atrial fibrillation (AF) leads to overload and disruption of the pumping function of the heart, which contributes to the development of heart failure; while a person experiences discomfort in everyday life [1].
The mechanisms of fibrillation have not been fully studied by the scientific world, especially in the case of structural changes in the heart (remodeling): myocardial infarction, myocarditis, cardiomyopathy, dilation, myocardial hypertrophy, heart defects, etc. In this article, we will offer you a new mechanism for the development of paroxysmal tachycardia, flutter and fibrillation of the heart, in comparison with the currently existing mechanism for the development of AF in structural changes in the heart [2].
“ANTIOXIDANT BARRIER”
Oxidative stress in the myocardium can be the cause of arrhythmia. This is due to the fact that oxidative stress disrupts the normal functioning of the heart muscle and electrical activity, which leads to heart rhythm disturbances. This is the conclusion given by artificial intelligence when searching in a neural network. This is an incorrect definition, oxidative stress should affect ectopic nodes that are separated from the myocardium by a connective tissue membrane, only then does arrhythmia occur.
Our body is not as simple as it may seem, any negative action will always be counteracted! The “antioxidant barrier” of the cardiac conduction pathways and ectopic nodes counteracts oxidative stress of the myocardium.
Now we will explain what the “antioxidant barrier” of the cardiac conduction pathways is and how it works. First, remember the following information:
- The cardiac conduction pathways start from the sinoatrial (SA) node and extend to the Pukinje fibers, almost like plant roots;
- The SA node, the atrioventricular (AV) node and all ectopic nodes located along the cardiac conduction pathways. Accordingly, the electrical impulse passes through all the nodes that are located on its path;
- An ectopic node cannot be located outside the cardiac conduction pathways, there are none in the myocardium;
- The electrical pathways of the heart and ectopic nodes (Figure 1) are separated from the myocardium by a connective tissue (insulating) sheath, approximately as in a regular wire (Figure 2); 5. Accordingly, only the Purkinje fibers have the right to conduct an electrical impulse to the myocardium.
.png)
Figure 1. Approximate location of SA, AV and other ectopic nodes of the heart (yellow circles along the conduction pathways of the heart).
In Figure 2, we have presented you with an approximate model of the distal electrical impulse pathway. Look at this picture and imagine that there is myocardium around the conduction pathway and oxidative stress has occurred.
.png)
Figure 2. Note: The figure shows an approximate model of the distal electrical conduction pathway.
The question is, how will the oxidation of the ectopic node and subsequent activation of arrhythmia occur? That's right, the weak link is the Purkinje fibers, through them the ectopic node will be oxidized!
Now look at Figure 3. A miracle has happened, antioxidant cells have appeared that will protect the ectopic node from oxidation. This is the “antioxidant barrier”, it consists of a connective tissue membrane and antioxidant cells.
.png)
Figure 3. Note: The figure shows an approximate model of the distal electrical conductive pathway.
Where did antioxidant cells come from? They were given to us by the great researcher Jan Evangelista Purkinje, he described his B-cells (later they were named in his honor - Purkinje fibers) and “transitional” T-cells [3]. Purkinje did not know the main purpose of T-cells and called them “transitional”, capable of conducting electricity from Purkinje fibers to the myocardium. But there is no sense in this, why is there a third conductor between two conductors of electricity?
We are sure that the main function of T-cells is to create an antioxidant barrier for the electrical pathways of the heart. At the time of the discovery of T-cells by the researcher Purkinje, the concept of “oxidative stress” of the myocardium did not exist. Then Purkinje would have understood that there is an “antioxidant barrier” of T-cells and connective tissue.
Accordingly, the conduction pathways of the heart have their own acid-base balance (pH). We don't know which one exactly, but we believe that it is T cells that regulate the electrolyte balance of the cardiac pathways (flows of Na+, Ca2+ and K+, etc.) (Figure 4).
.png)
Figure 4. Note: The figure shows a rough model of the “antioxidant barrier”, let's assume it is the Bachmann pathway. 2-5. Connective tissue sheath. 4. T cells. 1. Ectopic nodes (1 distal and 4 proximal), 3. Purkinje fibers.
Causes of damage to the "antioxidant barrier"
Structural pathology of the heart in cardiovascular diseases: myocardial infarction; sclerotic and post infarction heart failure; cardiomyopathy; heart defects with hypertrophy and dilation; cardiac surgery; myocarditis; pericarditis, etc. All these are pathological processes that lead to myocardial remodeling with possible damage to the electrical conductive pathways of the heart [4].
Structural changes in the heart can also be physiological, caused only by temporary but significant hemodynamic overload of large vessels that enter and exit the heart, such as: pulmonary veins; pulmonary artery; vena cava and aorta [5]. Due to hemodynamic overload of large vessels, myocardial overload occurs and the integrity of the antioxidant barrier is disrupted. For example, a patient has a severe bronchopulmonary disease, leading to hemodynamic overload in the pulmonary artery and the cavities of the right heart chambers, such a process can lead to excessive stretching of the myocardium and damage to the conductive pathways with the development of arrhythmia. Or with coarctation of the aorta, the pressure in it and its branches increases to the site of narrowing, and then decreases, this is the same factor of myocardial overload, with the probable development of damage to the conductive pathways and the antioxidant barrier.
Damage to the “antioxidant barrier” and the development of cardiac fibrillation (CF)
We do not divide the heart into atria and ventricles, damage to the conduction pathways of the heart can occur anywhere! But it is more dangerous if the damage is in the ventricles of the heart. When the “antioxidant barrier” is damaged at the level of T-cells or the lower sections of the connective tissue membrane, and also under conditions of oxidative stress, the intercellular fluid of the myocardium, rich in Na+ and K+ ions, penetrates into the distal ectopic nodes. Such hyperoxidation leads to irritation of the ectopic nodes and the development of FS [6] (Figure 5).
.png)
Figure 5. Damage to the necrotic zone of T cells that create an “antioxidant barrier” for the Purkinje fibers. Development of fibrillation. Note: the figure shows an approximate model of the electrical conduction pathway of the heart. 1. Connective tissue sheath and T cells. 2. Distal foci of ectopia (4 black dots). 3. Rotational waves of micro re-entry (red circular arrows). 4. Zone of myocardial necrosis (black dash).
Violation of the “antioxidant barrier” and the development of heart flutter (HF)
In some cases, damage to the connective tissue insulating sheath of the “antioxidant barrier” can be significant and localized in the proximal part of the conduction pathways. In this case, oxidation of one proximal ectopic node will occur, and most of the electrical impulse will go directly to the myocardium, bypassing the lower conduction pathways and Purkinje fibers. Such a process will form a large wave in the myocardium of macro re-entry and this will be - heart flutter (Figure 6). Only this mechanism explains the appearance of a large wave of macro re-entry [7].
.png)
Figure 6. Significant damage to the connective tissue (insulating) sheath in HF. Note: the figure shows an approximate model of the electrical conduction pathway of the heart. 1. Damage site - connective tissue sheath. 2. Proximal ectopic node (black dot). 3. Movement of a large macro re-entry wave (red circular arrow).
Accordingly, with the development of atrial flutter (AF), we will see large F waves on the electrocardiogram (Figure 7). Subsequently, if oxidation reaches the distal ectopic nodes, AF will turn into atrial fibrillation.
.png)
Figure 7. ECG, AF, correct shape, F waves.
Many researchers agree that paroxysmal supraventricular tachycardia (Figure 8) and AF are one type of arrhythmia. Pay attention to Figures 6 and 7, why such a difference? The explanation follows.
.png)
Figure 8. ECG, paroxysmal supraventricular tachycardia.
Damage to the “antioxidant barrier” and the development of paroxysmal tachycardia
Damage to the “antioxidant barrier” in the proximal conduction pathways may not be as significant as in the development of AF. These may be cracks or pores in the connective tissue sheath. In this case, only one ectopic node will be oxidized and paroxysmal supraventricular tachycardia will appear on the ECG, while the electrical impulse will go its own way, to the distal Purkinje fibers. With AF, the same thing happens, but instead of a crack in the connective tissue sheath, a large window of damage is formed. Accordingly, with AF, the electrical impulse will weakly reach the distal Purkinje fibers, most of it (90%) will immediately go to the myocardium through a large window of damage! Why should an electrical impulse move along conduction pathways that branch and narrow, have obstacles in the form of distal ectopic nodes, through which it also has to pass, when there is an open window of damage nearby!? On the ECG, this is displayed by large F waves.
You may ask why there is such a difference in heart rate: in paroxysmal supraventricular tachycardia, the atrial heart rate is up to 250 beats per minute, and in AF it is up to 350 beats per minute. About 100 beats per minute in favor of AF. This difference is due to the fact that the free release of an electrical impulse to the myocardium, through a large window of damage in AF, significantly accelerates the transmembrane action potential and shortens the resting potential than in paroxysmal tachycardia!
Accordingly, a small crack in the conduction pathway of the heart during the development of paroxysmal tachycardia always regenerates well, therefore this type of arrhythmia is never long-term, and even more so permanent, but if the damage increases, then paroxysmal tachycardia will turn into AF.
The existing mechanism of development of AF
Typically, premature atrial beats are the main triggers that convert sinus rhythm to AF. According to Haïssaguerre et al., the vast majority of these ectopic foci inside the atria originate in the pulmonary veins (PVs). The myocardial sleeves within the PVs appear to have specific properties that are both similar to and different from those of the rest of the atrial myocytes in terms of cellular electrophysiology, anatomical characteristics, and myofiber structure and orientation. The main aim of AF catheter ablation is the electrical isolation of the PVs from the rest of the atrium. This therapeutic strategy constitutes the cornerstone of AF catheter ablation [8]. Arrhythmogenesis has a predilection toward PV cardiomyocytes due to their action-potential characteristics, which renders them more susceptible to enhanced normal automaticity and triggered activity.
It is accepted that the acceleration of phase 4 depolarization results in enhanced normal automaticity, reaching an earlier threshold and an elevated automatic rate. On the other hand, delayed after-depolarization is the outcome of intracellular Ca+2 overload. In this process, he calcium-overloaded sarcoplasmic reticulum excretes Ca+2 in the diastole, activates Ca+2-dependent depolarizing currents (such as the Na+/Ca+2 exchange current) and, subsequently, produces a transient inward current that provokes membrane depolarization. The delayed after-depolarization reaches its threshold and a triggered ectopic action potential ensues. Early after-depolarizations originate from disproportionate action-potential prolongation caused by (a) the loss of repolarizing K+ currents, (b) excessive late components of Na+ currents and (c) the reactivation of plateau Ca+2 currents, which produce secondary arrhythmic depolarizations [9]. It is apparent that PV cells fulfill these criteria. The PVs have a depolarized resting membrane potential—which facilitates enhanced normal automaticity. Furthermore, PVs also provide an action potential with a lower amplitude, a shorter duration, and a smaller maximum phase 0 upstroke velocity. Slow and rapid delayed K+ rectifier currents are augmented in the PVs, whereas transient outward K+ currents and L-type Ca2+ currents are attenuated. Furthermore, animal studies have demonstrated that the diminished activity of the IK1 current facilitates trigger activity during the late phase of depolarization based on an afterdepolarization effect [10].
What is, however, the crucial stimulus that provokes the premature atrial beats in the pulmonary veins? The main AF comorbidities, such as hypertension, ischemic cardiomyopathy, diabetes mellitus, congestive heart failure, and advanced age alter left ventricular elastic properties and subsequently increase left atrial pressure [11]. The most common last step in this pathophysiologic cascade is the development of tissue stretch, which can also induce afterdepolarization and, thus, ectopic activity. This is facilitated by alterations in Ca+2 handling induced by increased Ca+2 excretion from the sacroplasmatic reticulum and enhanced Na+/Ca+2 exchange, particularly in the setting of b-adrenergic stimulation. Additionally, angiotensin II contributes similarly to Ca+2-handling changes by activating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which is a significant downstream effector in cellular oxidative stress. It is known that NADPH oxidase generates reactive oxygen species (ROS). In turn, ROS products induce early afterdepolarization (EAD) and delayed afterdepolarization (DAD) as a trigger activity through the enhancement of late Na+ currents. Angiotensin II and ROS enhance abnormal Ca+2 handling and Ca+2 overload, which cause increased atrial contractility and upregulate Ca+2-dependent signaling. The subsequent alteration of the intracellular calcium balance encourages early after-depolarization. The Ca+2-induced activation of the nuclear factor of activated T cells (NFAT) suppresses the L-type Ca+2-current function, decreases action-potential duration (APD), and facilitates AF-induced electrical remodeling [12] (Figure 9).
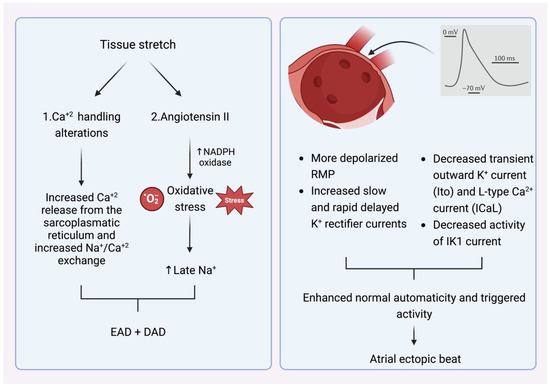
Figure 9. (Left) Mechanism that explains how tissue stretch promotes atrial premature beats, mainly through afterdepolarization. (Right) Specific electrophysiologic properties of the pulmonary veins render them more likely to be the origin of atrial ectopic beats. NADPH oxidase (nicotinamide adenine dinucleotide phosphate oxidase) EAD: early afterdepolarization. DAD: delayed afterdepolarization. RMP: resting membrane potential.
Triggers may originate from different regions (non-PV triggers), including the superior vena cava, crista terminalis, coronary sinus, left atrial appendage, left atrial posterior free wall, and ligament of Marshall [13]. Specific mapping protocols and isoprenaline have been used to address the significance of inducing, locating, and eliminating such non-PV triggers as a means to achieving better results in AF ablation in comparison to empirical PVI. However, this therapeutic strategy has not been established as a common practice [14] (Figure 10).
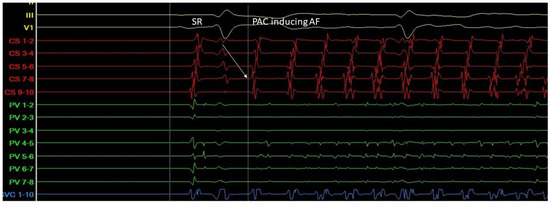
Figure 10. A. Atrial ectopic beat originates from the proximal aspect of the coronary sinus (arrow at the CS 7-8 electrogram)—a non PV trigger—and induces atrial fibrillation (G. Leventopoulos’ archive). PV: Pulmonary Vein. CS: Coronary Sinus.
CONCLUSION
Only a deep and complete understanding of the mechanisms involved in the development of cardiac fibrillation will allow us to develop more specific prevention and treatment of this debilitating disease, and sometimes death. There are other mechanisms for the development of cardiac fibrillation, for example, with weakness of the SA node, prolonged activity of the sympathoadrenal or cholinergic systems, etc. Unfortunately, the mechanism for the development of cardiac tremor and fibrillation that we have proposed will not answer all the theoretical mechanisms for the development of these arrhythmias. But we have defined a theory related to damage to the "antioxidant barrier" of the cardiac conduction pathways during structural changes in the myocardium.
Thus, if we assume that a small rupture of the insulating sheath of the conduction pathway can occur, as shown in Figure 11.

Figure 11. A small rupture of the connective tissue (insulating) sheath.
This damage can heal itself during the scarring period, but paroxysmal and persistent forms of AF will be observed. If the damage to the insulating sheath of the conduction pathway is more serious, as in Figure 12, then AF will be long-term - a permanent form.

Figure 12. Significant rupture of the connective tissue (insulating) sheath.
We believe that patients with newly developed paroxysmal and persistent forms of AF should be given reparative and antiacidemic therapy to accelerate tissue regeneration in damaged areas of the heart, which will help the body restore the integrity of the cardiac conduction pathways.
AUTHOR CONTRIBUTIONS
All authors meet the ICMJE criteria for authorship. Author contributions (according to the Credit system): Khabchabov R.G. – article concept, source search, manuscript creation and editing, approval of the final version of the article; Makhmudova E.R. – article concept, source search, manuscript creation and editing, approval of the final version of the article.
ACKNOWLEDGEMENTS
None.
CONFLICT OF INTEREST
The authors declare no obvious or potential conflicts of interest or personal relationships related to the publication of this article. The authors did not declare any other conflicts of interest.
FUNDING FOR THE ARTICLE
None.
REFERENCES
- Stefil М, Lip GYH. (2022). Аtrial fibrillation. Arrhythmias and electrophysiology. 50(8):516-521.
- Bockeria LA, Shengelia LD. (2014). Mechanisms of atrial fibrillation: from ideas and hypotheses to effective understanding of the problem. Annaly Aritmologia. 11(1):4-14.
- Wang Y, Hill JA. (2010). Electrophysiological Remodeling in Heart Failure. J Mol Cell Cardiol. 48(4):619-632.
- Jansen HJ, Bohne LJ, Gillis AM, Rose RA. (2020). Atrial remodeling and atrial fibrillation in acquired forms of cardiovascular disease. Heart Rhythm O2. 1(2):147-159.
- Kim HJ. (2020). Clinical Implications of Changes in Cardiac Structure and Function after Extreme Endurance Exercise. J Cardiovasc Imaging. 28(3):211-212.
- Khabchabov RG, Makhmudova ER, Dzhanbulatov MA. (2021). Effect of antiacidemic therapy on arrhythmia in patients with acute myocardial infarction after percutaneous transluminal coronary intervention. Actual problems of medicine. 44:174-182.
- Khabkhabov RG, Makhmudova ER. (2016). Antiarrhythmic effect of antioxidants in patients with atrial fibrillation. J Atr Fibrillation. 8(6):40-44.
- Haïssaguerre M, Jaïs P, Shah DC, Takahashi A, Hocini M, Quiniou G, et al. (1998). Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 339(10):659-666.
- Nattel S. (2003). Atrial Electrophysiology and Mechanisms of Atrial Fibrillation. J. Cardiovasc. Pharmacol. Ther. 8(Suppl. 1):S5-S11.
- Ehrlich JR, Cha TJ, Zhang L, Chartier D, Melnyk P, Hohnloser SH, et al. (2003). Cellular electrophysiology of canine pulmonary vein cardiomyocytes: action potential and ionic current properties. J Physiol. 551(Pt 3):801-813.
- Weber KT, Brilla CG, Campbell SE, Guarda E, Zhou G, Sriram K. (1993). Myocardial fibrosis: role of angiotensin II and aldosterone. Basic Res Cardiol. 88(Suppl 1):107-124.
- Harada M, Van Wagoner DR, Nattel S. (2015). Role of inflammation in atrial fibrillation pathophysiology and management. Circ J. 79(3):495-502.
- Voigt N, Dobrev D. (2013). The biology of human pulmonary veins: does it help us to better understand AF pathophysiology in patients? Heart Rhythm. 10(3):392-393.
- Santangeli P, Marchlinski FE. (2017). Techniques for the provocation, localization, and ablation of non-pulmonary vein triggers for atrial fibrillation. Heart Rhythm. 14(7):1087-1096.