Information Links
Related Conferences
Previous Issues Volume 8, Issue 1 - 2024
Study of Key Aspects of Anticoagulant and Antiplatelet Therapies: New Perspectives and Innovations in Assessing the Coagulation System's State
Andrey Belousov1,2*, Ekateryna Belousova1
1Laboratory of Applied Nanotechnology of Belousov, Ukraine
2Kharkiv National Medical University, Ukraine
*Corresponding Author: Dr. Andrey Belousov, Laboratory of Applied Nanotechnology of Belousov, Kharkov Medical, Academy of Postgraduate Education, Ukraine, Website: www.nanolab.com.ua; E-mail: [email protected].
Received Date: January 04, 2024
Published Date: February 06, 2024
Citation: Belousov A, et al. (2024). Study of Key Aspects of Anticoagulant and Antiplatelet Therapies: New Perspectives and Innovations in Assessing the Coagulation System's State. Mathews J Cardiol. 8(1):32.
Copyrights: Belousov A, et al. © (2024).
ABSTRACT
Statement of the Problem: Impairment of the blood coagulation system (BCS) remains a relevant issue in clinical practice. A striking example of this is the fact that despite the administration of anticoagulant therapy, up to 35% of COVID-19 patients were hospitalized in the intensive care unit with thromboembolic complications. Prophylactic and therapeutic methods for the hemostatic system, recommended by various protocols, have a general nature. Moreover, they introduce dissonance into the clinical clarity of appropriate anticoagulant and antiplatelet use and fail to provide practitioners with the necessary fundamental knowledge to understand the aspects of clinical tactics for correcting the hemostatic system. Uncertainties remain regarding how to correctly choose a specific anticoagulant and determine its effective dose based on individual clinical and laboratory data, which laboratory markers of the coagulation system should be investigated, and whether they always reflect the true picture of hemostasis. Methodology and Theoretical Orientation: This article briefly presents the main aspects of the functioning of the BCS, points of application for pharmacological agents, and provides objective information for clinicians about the mechanisms of action of major anticoagulants and antiplatelets. Effective and practical methods for assessing the hemostatic system are recommended. Findings: The proper correction of the hemostatic system can only be carried out by a highly qualified specialist - a clinical transfusiologist, who possesses a comprehensive understanding of the functioning of the coagulation and anticoagulation systems, the platelet and fibrinolysis systems, knows the points of application and mechanisms of action of the pharmacological agents and blood components used, and takes into account internal and external factors that influence hemostasis. Conclusion and Significance: Empirical prescription of anticoagulant and antiplatelet therapies, based solely on instructions and clinical protocols without objective comprehensive analysis of the hemostatic components, is not only ineffective but also life-threatening for the patient. Coagulogram and D-dimer measurements cannot serve as the sole determining markers for monitoring the coagulation system. Evaluation of the BCS should only be carried out based on a comprehensive analysis of coagulogram data, complete blood count, thromboelastography (TEG), and data from all relevant systems.
Keywords: Anticoagulants, Antiplatelets, Thrombosis, Hemostasis Assessment, Regulation.
STATEMENT OF THE PROBLEM
Despite the availability of a wide variety of anticoagulants and antiplatelet agents in the market, the impairment of the BCS remains a relevant issue in clinical practice. One example of this is the numerous publications dedicated to the novel coronavirus infection (COVID-19), which, despite the administration of anticoagulant therapy, often leads to systemic activation of blood coagulation and thrombotic complications [1-4]. Initial studies of COVID-19 patients demonstrated a high frequency of venous thromboembolism, including pulmonary embolism (PE). Despite receiving anticoagulant therapy, up to 35% of COVID-19 patients required admission to the intensive care unit due to thromboembolic complications. Severe complications of COVID-19 were associated with disturbances in coagulation processes and characterized by elevated levels of D-dimers and fibrinogen. An increased risk of developing PE was observed, which directly contributed to the death of approximately 30% of patients with COVID-19 [5].
In a prospective observational study by J. Helms et al., PE was observed in 16.7% of patients despite receiving anticoagulant therapy [6]. Interestingly, there were no signs of deep vein thrombosis or other sources of thromboembolism. According to the findings of R. Beun et al. (Netherlands), among 75 patients admitted to the intensive care unit (ICU), the incidence of PE was 26.6% [7]. In the study by J. Poissy et al., out of 107 patients hospitalized in the ICU and receiving anticoagulant therapy, 22 (20.6%) developed PE [8]. In a retrospective cohort study conducted by F.A. Klok et al., among 184 patients admitted to the ICU, PE occurred in 13.6% of patients despite receiving anticoagulant therapy [9]. According to a systematic review and meta-analysis by L. Roncon et al., conducted on a sample of 7178 COVID-19 patients, the overall frequency of thromboembolic complications (pulmonary embolism) among those hospitalized in general wards and ICUs was 14.7% and 23.4%, respectively [10].
Currently, several different pathophysiological mechanisms are considered to contribute to the development of PE in patients with COVID-19, including increased systemic inflammatory response, disruptions in the hemostatic system, the development of pulmonary intravascular coagulopathy, and endothelial dysfunction [11]. A study by H. Han et al. conducted at Wuhan University (China, 2020) confirmed the development of a hypercoagulable state in patients with COVID-19, characterized by elevated levels of D-dimers, fibrinogen, and fibrinogen degradation products [12]. M. Oudkerk et al. suggest that the high level of D-dimers observed in COVID-19 patients is a consequence of systemic inflammation and reflects the true thrombotic state induced by the virus [13]. Sakr Y. et al. (2020) believe that the thrombosis occurring in COVID-19 patients despite anticoagulant therapy is a result of insufficient data on the epidemiology and pathophysiological mechanisms obtained from randomized clinical trials [14]. Meanwhile, Z. Zhai et al. (2020) recommend the use of low molecular weight heparins (LMWH) as first-line therapy for patients with mild or moderate COVID-19 who have a high or moderate risk of developing PE. The duration of prophylactic therapy should be at least 7-10 days or continued until the elimination of venous thrombosis risk factors [15].
The American Society of Hematology (ASH) and the International Society on Thrombosis and Haemostasis (ISTH) recommend the administration of prophylactic doses of LMWH (4000 IU twice daily) to all hospitalized patients with confirmed diagnosis of COVID-19. Pharmacological prophylaxis with LMWH is recommended as the first-line approach for patients with low or moderate risk of bleeding and no contraindications to antithrombotic agents. In cases of severe renal insufficiency (creatinine clearance <30 mL/min), unfractionated heparin (UFH) is recommended [16-18].
The choice of the appropriate medication and correct dosage requires taking into account comorbidities such as renal or hepatic dysfunction, thrombocytopenia, and gastrointestinal conditions. As a first-line therapy, Z. Zhai et al. recommend the use of parenteral LMWH, such as enoxaparin (100 IU/kg twice daily) or enoxaparin (150 IU/kg once daily) or nadroparin (86 IU/kg twice daily) [15]. The examples provided in the given information create dissonance and confusion in the clinical clarity of the proper use of anticoagulants and antiplatelet agents, as it fails to provide healthcare professionals with the necessary fundamental knowledge to choose the correct strategy for correcting the hemostatic system. Several important questions arise:
- How to correctly choose a medication and determine its effective dosage considering individual clinical and laboratory data of the patient?
- Which laboratory and instrumental methods of investigation reflect the true state of hemostasis?
- Is it accurate to assess the hemostatic condition based solely on coagulation profile and D-dimer levels?
These questions highlight the need for providing objective information to clinicians in order to ensure the proper and effective use of anticoagulants and antiplatelet agents.
In clinical practice, drugs that affect the BCS are widely used by physicians of various specialties, often as part of comprehensive therapy and symptomatic treatment.
However, in order to effectively and appropriately utilize such medications, it is imperative to possess profound knowledge regarding the functioning of the coagulation and anticoagulation systems, fibrinolysis mechanisms, the influence of the body's internal environment in maintaining their balance, and a comprehensive understanding of the overall pharmacological effects of the drugs employed. Proper use of hemostatic agents is impossible without adequate assessment of the BCS and a balanced, comprehensive approach to therapy. Ignoring this fact increases the risk of serious complications related to disruptions in the blood coagulation system, which can be life-threatening for the patient.
The objective of this study is to provide clinicians with objective information on the mechanisms of action of major types of anticoagulants and antiplatelet agents, based on fundamental knowledge of the functioning of the blood coagulation system. Additionally, the study aims to recommend effective methods for assessing the state of the hemostatic system.
METHODOLOGY AND THEORETICAL ORIENTATION
Factors Influencing the Development of Venous Thrombosis. Key Causes of Thrombosis in COVID-19
The occurrence of venous thrombosis can be attributed to various factors, which are encompassed by Virchow's triad [16]:
- Reduced blood flow velocity;
- Endothelial dysfunction;
- Activation of the blood coagulation system.
With the emergence of COVID-19, Sakr Y. et al. (2020) presented their concept of hypercoagulability pathogenesis in this disease [17]. The main causes of blood coagulation abnormalities are as follows:
- Severe hypoxia, associated comorbidities, and organ dysfunction predispose individuals to hemostatic disturbances. Hypoxia increases blood viscosity, thereby promoting thrombosis.
- Endothelial dysfunction, elevation of von Willebrand factor, toll-like receptor activation, and tissue factor activation can induce proinflammatory and procoagulant effects through complement activation and cytokine release. This leads to dysregulation of the coagulation cascade, resulting in the formation of pulmonary intravascular or systemic thrombi.
- The "cytokine storm" induces secondary development of hemophagocytic lymphohistiocytosis with coagulation activation, increasing the risk of intravascular microthrombosis and secondary coagulopathy, which contribute to the occurrence of thromboembolism.
It should be noted that the presented list of thrombogenesis causes in COVID-19 by Sakr Y. et al. is pathogenetically predictable and fully aligns with the classical Virchow's triad. The development of systemic endothelial dysfunction in COVID-19 accounts for the loss of certain physiological properties of the endothelium, including its ability to stimulate vasodilation, fibrinolysis, and antiplatelet aggregation. Additionally, viral and cytokine-induced endothelial cell damage leads to excessive release of von Willebrand factor, triggering thrombus formation. Consequently, CT-pulmonary angiography reveals widespread cases of obliteration and thrombosis in small and medium-sized vessels, which are secondary manifestations of COVID-19 [18].
The Peculiarities of Using Various Types of Anticoagulants and Their Points of Application
The implementation of anticoagulant therapy and the selection of anticoagulants should be based not on recommendations derived from controversial statistical data analysis of meta-analyses, subjective and constantly changing treatment protocols, but on objective, fundamental knowledge about the functioning of the hemostatic system, the pharmacological mechanisms of action of the drugs, and a personalized approach to assessing the state of the coagulation system.
Let us begin by examining the points of application of major types of anticoagulants. The application points of major types of anticoagulants and their mechanisms of action are illustrated in Figures 1-4, based on the blood coagulation scheme developed by Paul Morawitz (1905) [19,20].
.png)
Figure 1. Points of application of indirect and direct anticoagulants.
Figure 1 illustrates that indirect anticoagulants inhibit the activation of prothrombin (II), proconvertin (VII), antihemophilic globulin B (IX), and thrombotropin (X) by suppressing the reduction of Vitamin K1 epoxide, which are synthesized in the liver and play a crucial role in the formation of fibrin clots [21]. However, it should be noted that simultaneously, antagonists of Vitamin K hinder the activation of proteins C (XIVa) and S, which possess anticoagulant activity and are also synthesized in the liver. In light of this, practicing physicians should pay attention to the danger of hypercoagulation occurring within the first three days after initiating the administration of indirect anticoagulants (especially neodicoumarin). This is due to the fact that when indirect anticoagulants are prescribed, there is a more rapid decrease in the levels of proteins C and S, which have a shorter half-life compared to factors II, VII, IX, and X.
Therefore, in acute situations (such as myocardial infarction, etc.), it is essential to administer heparin concurrently with Vitamin K antagonists in the first three days as a preventive measure against thrombosis. Ignoring this can lead to a decrease in the levels of proteins C and S, which can cause the development of so-called "coumarin" necroses in soft tissues (buttocks, mammary glands, cheeks, genitalia), resulting from capillary and small venule thrombosis. These necroses typically appear 4-10 days after starting the medication, more frequently in women. To address the onset of complications, transfusion of frozen blood plasma rich in protein C is performed [22].
Also, Figure 1 shows the points of action of unfractionated heparins (UFH) and low molecular weight heparins (LMWH). The difference in their action lies in the fact that UFH inhibits the activation of plasma factors II and X, while LMWH primarily inhibits the activation of factor X. Importantly, all heparins act indirectly on plasma factors through a protein mediator called antithrombin III. Antithrombin III is an anticoagulant protein, with the average reference values of normal ranging from 80% to 120%. Antithrombin III inactivates coagulation factors (IIa, VIIa, XIa, Xa, IXa, XIIa). Specific regions of heparins bind to the positively charged amino groups in the antithrombin III molecule. As a result, the reactivity of arginyl groups in antithrombin III increases. Interacting with the active sites of serine proteases (factors IIa, VIIa, IXa, Xa, XIa, XIIa), arginine groups of antithrombin III inhibit the activity of plasma factors and prevent the formation of fibrin clots [23, 24]. Antithrombin III is primarily synthesized by the liver and vascular endothelium.
Therefore, practicing physicians should understand that in the presence of endothelial dysfunction and signs of liver dysfunction, situations may arise where the level of antithrombin III falls below 80%. In such cases, the use of heparins may become ineffective in the presence of clinical and laboratory signs of thrombosis. Thus, it becomes crucial and necessary to determine the level of antithrombin III in the blood through laboratory testing. If the level of antithrombin III is below 80%, the administration of heparins is not indicated. The level of antithrombin III can be increased through the transfusion of human antithrombin III concentrate ("Atenativ") [25].
Another important aspect that clinicians should consider is that heparins belong to the class of proteoglycans. In response to their administration, the immune system can activate the production of antibodies against the heparin-calcium complex. This leads to the development of an immune type of heparin-induced thrombocytopenia (Figure 2). In immune type of heparin-induced thrombocytopenia, increased platelet aggregation occurs on days 9-14, along with thrombocytopenia (less than 50% of the baseline), decreased blood clotting time, and the presence of antibodies against the heparin-calcium complex in the blood [26]. The inability of clinicians to diagnose immune type of heparin-induced thrombocytopenia at early stages leads to erroneous tactics of increasing the dosage of heparins. Ultimately, this becomes the main cause of "rebound" thrombosis. It should be noted that the risk of developing "rebound" thrombosis is very high in patients with COVID-19. This is due to the fact that in many patients, viral-induced endothelial damage triggers an autoimmune inflammatory process [27]. Consequently, in the presence of heparin administration, typically on days 9-14, the clinical manifestation of venous thrombosis or pulmonary embolism rapidly occurs. In the event of clinical and laboratory signs of immune type of heparin-induced thrombocytopenia, heparin therapy should be immediately discontinued and replaced with fondaparinux. Fondaparinux belongs to the class of pentasaccharides and does not induce immune type of heparin-induced thrombocytopenia [28]. However, it should be noted that fondaparinux, similar to heparins, exerts its effects through antithrombin III, but unlike heparin, it exhibits selective action towards factor Xa.
.png)
Figure 2. Development of heparin-induced thrombocytopenia in response to the administration of heparins.
Figure 2 demonstrates that the action of heparins and fondaparinux requires antithrombin III [29]. In cases where it is not possible to determine the level of antithrombin III or if it is low, the following anticoagulants are prescribed - rivaroxaban, apixaban, and dabigatran.
Figure 3 complements the points of application of these anticoagulants.
.png)
Figure 3. Points of application of rivaroxaban, apixaban, and dabigatran.
Unlike heparins and fondaparinux, these anticoagulants directly inhibit activated plasma coagulation factors without the need for the protein mediator antithrombin III [30]. The difference in their action lies only in the fact that rivaroxaban and apixaban target factor Xa, while dabigatran targets factor IIa. For the appropriate use of anticoagulants, clinicians should also have knowledge of the function of protein C. Protein C is an anticoagulant protein. Under the action of thrombomodulin on the surface of endothelial cells, protein C is converted into an active protease and, after interaction with protein S, exhibits pronounced anticoagulant activity. The main function of activated protein C is the hydrolysis of plasma factors Va and VIIIa, preventing the chain reaction of thrombin and fibrin formation [31]. For example, one of the causes of recurrent venous thrombosis in 20-50% of cases is the mutation in factor V (known as Leiden mutation) [32]. In this case, the mutated factor V becomes resistant to hydrolysis by protein C. Therefore, clinicians must consider this fact when deciding on the discontinuation of anticoagulants.
Laboratory Screening and Monitoring of Anticoagulant Therapy
The screening and monitoring of antithrombotic therapy are presented in Table 1.
Table 1. Laboratory Screening and Monitoring of Antithrombotic Therapy
|
Anticoagulants |
Points of application |
Laboratory indicator |
|
UFH |
IIa, Xa |
Activated partial thromboplastin time (APTT) |
|
LMWH |
IIa, Xa |
Anti Xa |
|
Fondaparinux |
Xa |
Anti Xa |
|
Indirect anticoagulants |
(II,VII,IX,X,XIV)а |
Prothrombin time (PT), international normalized ratio (INR) |
|
Rivaroxaban |
Xa |
PT (Neoplastin kit), anti- Xa calibration test |
|
Apixaban |
Xa |
PT (Neoplastin kit), anti- Xa calibration test |
|
Dabigatran |
IIa |
APTT, Thrombin time (TT) |
Key Aspects of the Application of Various Antiplatelet Agents and Their Points of Application
Antiplatelet agents are primarily used for the prevention and treatment of arterial thrombosis. The points of application for major types of antiplatelet agents are presented in Table 2.
Table 2. Points of Application for Major Types of Antiplatelet Agents
|
Anticoagulants |
||
|
№ |
Points of application |
Medications |
|
1 |
Cyclooxygenase 1,2 inhibitors |
Acetylsalicylic acid (Aspirin) |
|
2 |
Phosphodiesterase cAMP inhibitors |
Dipyridamole (Courantil) |
|
Pentoxifylline (Trental) |
||
|
Cilostazol (Pletal) |
||
|
3 |
ADP receptor antagonists |
Clopidagrel |
|
Ticaglerol |
||
|
Prasugrel |
||
|
4 |
GPIIb/IIIa receptor antagonists |
|
|
4.1 |
Monoclonal antibodies: |
|
|
Abcisquimab (Reo-Pro) |
||
|
Monafram (Framon) |
||
|
4.2 |
Cyclic peptides: |
|
|
Eptifibatide (Integrilin) |
||
|
Non-peptide blockers: |
||
|
4.3 |
Tirofiban (Agrostat) |
|
|
Orphobitan |
||
|
Xemilofiban |
||
|
Lamifiban |
||
|
Sibrafiban |
||
Typically, when deciding on the prescription of antiplatelet therapy, clinicians rely on clinical data, protocols, and pharmacological instructions for use. However, they do not consider the actual activity state of different types of receptors on the surface of platelets, nor do they objectively monitor their functions. Ultimately, this not only contributes to the low effectiveness of antiplatelet therapy but also becomes the main cause of life-threatening conditions for the patients.
To address this issue, clinicians need to choose an effective and practical method for the objective assessment and monitoring of platelet aggregation function. One such method is Thromboelastography (TEG). TEG allows for rapid and efficient monitoring of the state of key factors in the blood coagulation system. This method can provide an objective assessment of platelet aggregation function and enable clinicians to dynamically observe the measured parameters. Additionally, TEG can easily and quickly evaluate the effectiveness of different antiplatelet agents. However, it is important to note that the interpretation of TEG data should be performed by a qualified physician, specifically a clinical transfusion specialist.
Among all the types of antiplatelet agents used, particular attention should be given to the mechanism of action of acetylsalicylic acid, which is commonly prescribed by practicing physicians for the prevention and treatment of arterial thrombosis, according to widely used protocols and recommendations [34]. Clinicians should be aware that the mechanism of action of acetylsalicylic acid is not straightforward. The pharmaceutical manufacturers provide only one aspect of the mechanism of action of acetylsalicylic acid in the product instructions, which is the inhibition of cyclooxygenases (COX), leading to a reduction in thromboxane A2 synthesis in platelets. In turn, thromboxane A2 stimulates the activation of new platelets and their aggregation by increasing the expression level of the glycoprotein complex GP IIb/IIIa on the platelet membranes. Circulating plasma fibrinogen binds to this complex, thereby strengthening the thrombus [35]. It is believed that platelets only contain COX-1 [36]. Therefore, COX-1 inhibitors reduce the synthesis of thromboxane A2 in platelets. Ultimately, this reduces the activity of thrombus formation. It was this one-sided spectrum of action that was the determining factor in prescribing acetylsalicylic acid.
However, it should be noted that acetylsalicylic acid does not belong to selective COX-1 inhibitors. In addition to COX-1, it inhibits COX-2 as well [35]. COX-2 synthesis occurs in macrophages, monocytes, fibroblasts, chondrocytes, and endothelial cells. As a result of COX-2 inhibition, the synthesis of prostacyclin is reduced. This contributes to the formation of arterial thrombi and increases the risk of thrombosis in patients in the "at-risk" group [37]. This mechanism also underlies the pathogenesis of reducing inflammation, pain, and fever when using acetylsalicylic acid. Many authors in their scientific publications, using traditional nonsteroidal anti-inflammatory drugs (NSAIDs) as an example, claim that simultaneous inhibition of the two isoenzymes, COX-1 and COX-2, does not disrupt the balance from a hemostatic perspective [38]. In this regard, a logical question arises: how does acetylsalicylic acid, which simultaneously inhibits both isoenzymes (COX-1 and COX-2), selectively provide an antiplatelet effect? A meta-analysis of data from 16 trials involving 171,215 individuals with an average age of 64 years revealed new evidence that casts doubt on the usefulness and effectiveness of low-dose aspirin for the prevention of cardiovascular events. It was shown that in the majority of patients who regularly took acetylsalicylic acid, the antiplatelet effect was completely absent [39,40].
Such studies are not new. Previously, there has been a hypothesis that there are patients who are genetically insensitive to the action of acetylsalicylic acid. This hypothesis may only partially hold true. The balance of hemostasis depends not only on the activity of the drug to inhibit COX-1 and COX-2 but also on the quantitative content of polyunsaturated fatty acids (PUFAs), which are components of cell membrane phospholipids. Two classes of PUFAs are of practical interest to clinicians - omega-3 and omega-6. There are two main metabolic pathways for PUFAs - cyclooxygenase and lipoxygenase pathways (see Figure 4).
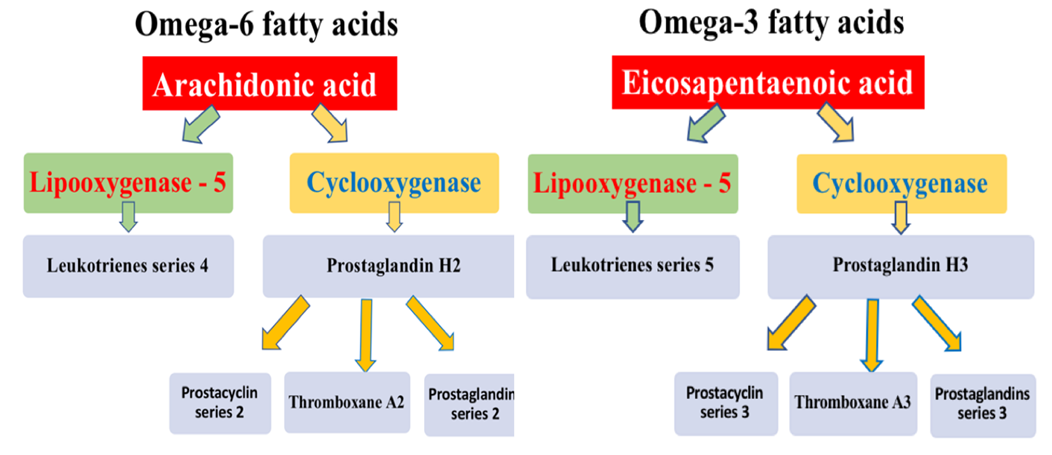
Figure 4. Scheme of eicosanoid synthesis from omega-6 and omega-3 PUFAs.
Figure 4 demonstrates that the cyclooxygenase pathway of PUFA metabolism leads to the formation of prostaglandins, prostacyclin, and thromboxane. The lipoxygenase pathway leads to the formation of leukotrienes. The key representative of omega-6 PUFAs is arachidonic acid (AA). Arachidonic acid is a polyunsaturated fatty acid present in the phospholipids (especially phosphatidylethanolamine, phosphatidylcholine, and phosphatidylinositides) of membranes of the body's cells, and is abundant in the brain, muscles, and liver. The cyclooxygenase pathway of AA metabolism results in the formation of prostaglandins and prostacyclin of series 2, as well as thromboxane A2. The lipoxygenase pathway leads to the formation of leukotrienes of series 4. The key representative of omega-3 PUFAs is eicosapentaenoic acid (EPA). The cyclooxygenase pathway of EPA metabolism leads to the formation of prostaglandins and prostacyclin of series 3, as well as thromboxane A3. The lipoxygenase pathway leads to the formation of leukotrienes of series 5 [41].
The functional properties of eicosanoids synthesized from omega-6 and omega-3 PUFAs are diametrically opposite. Prostaglandins and prostacyclin of series 3, derived from omega-3 PUFAs, promote platelet disaggregation and exert vasodilatory effects. In contrast, prostaglandins and prostacyclin of series 2, synthesized from omega-6 PUFAs, inhibit platelet disaggregation and induce vasoconstriction. It should also be noted that prostaglandins are formed in virtually all organs and tissues. Thromboxane A3, synthesized from omega-3 PUFAs, exhibits anti-aggregatory effects. Conversely, thromboxane A2, synthesized from omega-6 PUFAs, activates platelet aggregation. Similar differences are observed in the synthesis of leukotrienes. Leukotrienes of series 5, synthesized from omega-3 PUFAs, exert pronounced anti-inflammatory effects. Meanwhile, leukotrienes of series 4, synthesized from omega-6 PUFAs, potentiate inflammation [42]. On average, the omega-3/omega-6 ratio in the human body is 1:4 [43].
However, it should be noted that in certain tissues, the omega-3/omega-6 ratio shifts towards an increase in omega-3 polyunsaturated fatty acids (PUFAs). For instance, in the cells of the brain and hepatocytes, this ratio is 1:2 [44-47].
Taking the above into consideration, it becomes clear that the clinical effects of acetylsalicylic acid and other NSAIDs directly depend on the initial ratio of PUFA quantities present in cell membrane phospholipids. This fact is the main reason for the paradoxical clinical effects observed with the use of NSAIDs and must be taken into account by clinicians. Furthermore, for the effective and safe use of COX inhibitors, it is essential to have knowledge of the comprehensive mechanism of action of NSAIDs, rather than relying solely on pharmacological instructions, recommendations, and protocols. The selection and monitoring of anticoagulant and antiplatelet therapies should be based on comprehensive data from laboratory and instrumental research methods, enabling the efficient and safe correction of the hemostatic system.
One of the Common Methods Used in Clinical Practice to Assess the Hemostatic System is Coagulogram
Clinicians often attempt to evaluate the state of hemostasis based solely on coagulogram parameters and use pharmacological agents to correct it. However, this laboratory method of monitoring the BCS primarily reflects the potential capabilities of plasma factors to support hemostasis and often does not reflect their implementation in action.
Therefore, an analysis of the hemostatic system based solely on coagulogram data cannot fully characterize the true state of the blood coagulation system.
An effective method that allows for a rapid assessment of the implementation of main components of the BCS is thromboelastography (TEG) [48]. Unlike the coagulogram, TEG is an instrumental laboratory diagnostic method that reflects the process of thrombus formation and fibrinolysis in real-time. The interpretation of TEG data allows for an objective evaluation of the state of the plasma and platelet components of hemostasis, as well as the fibrinolysis system. The confirmation of the above is supported by common clinical examples of discrepancies between coagulogram data and thromboelastography (TEG) parameters, as illustrated in Figures 5-8.
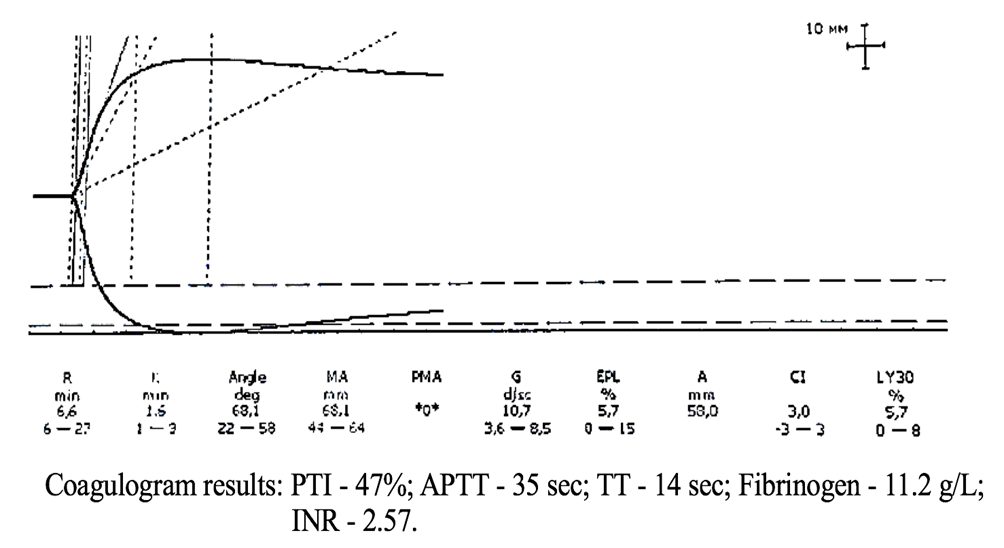
Figure 5. TEG parameters against the background of coagulogram data of patient K.
The coagulogram data presented in Figure 5 indicate pronounced hypocoagulation (high INR values and low PTI) in the presence of hyperfibrinogenemia. In contrast, the thromboelastogram (TEG) objectively registers a high risk of hypercoagulation due to hyperfibrinogenemia.
Another clinical case scenario is presented in Figure 6.
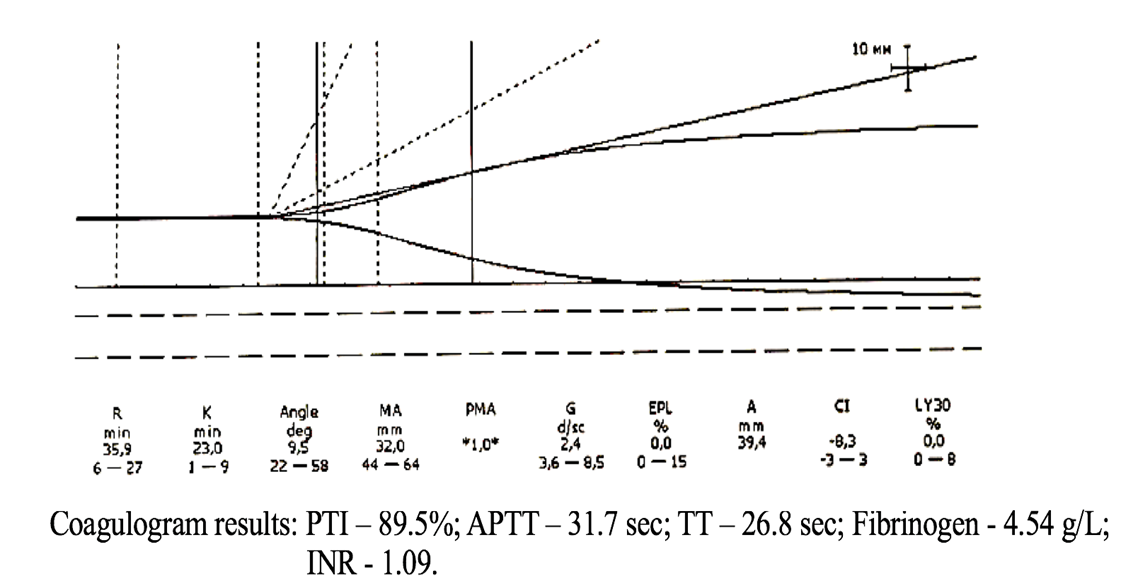
Figure 6. TEG parameters against the background of coagulogram data of patient R.
In Figure 6, the coagulogram data only indicate a risk of hypocoagulation. At the same time, the thromboelastogram (TEG) shows pronounced hypocoagulation that requires immediate correction.
The next clinical case scenario is presented in Figure 7.
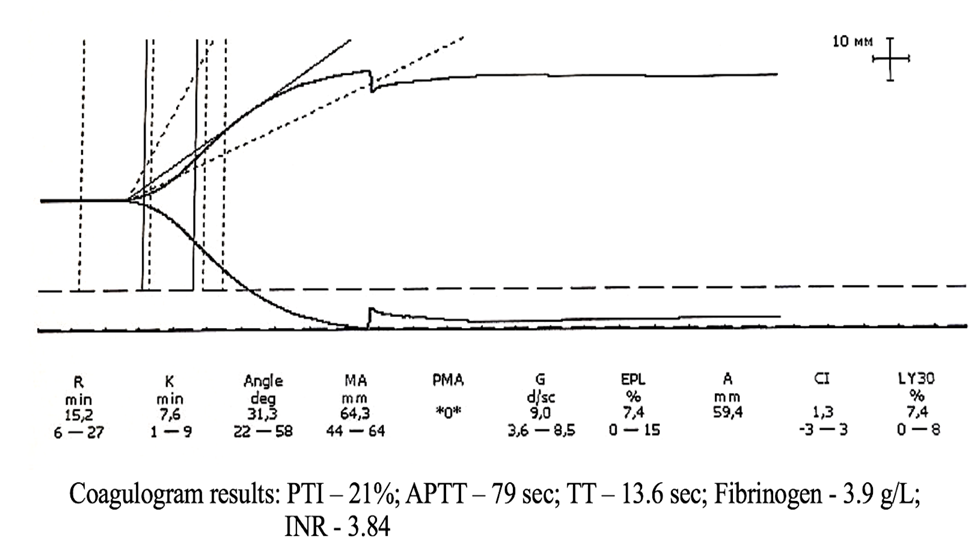
Figure 7. TEG parameters against the background of coagulogram data of patient M.
In Figure 7, the coagulogram parameters indicate pronounced signs of hypocoagulation that require urgent pharmacological correction. Conversely, the thromboelastogram (TEG) data demonstrate increased platelet aggregation. However, the hemostatic system remains balanced.
The fourth clinical example of TEG and coagulogram parameters is presented in Figure 8.
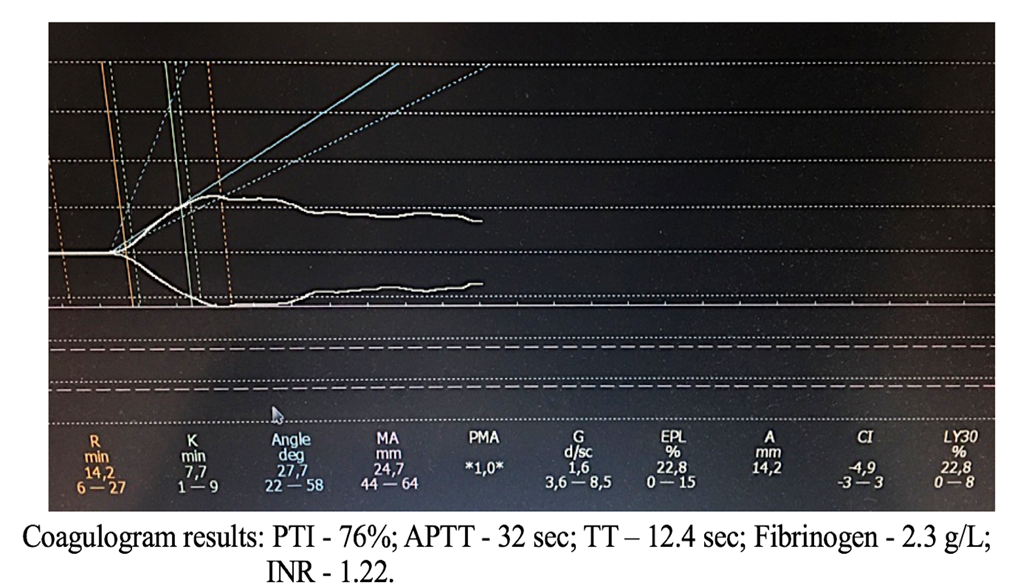
Figure 8. TEG parameters against the background of coagulogram data of patient S.
Figure 8 demonstrates that the coagulogram parameters are mostly within the normal range. However, the TEG clearly registers a decrease in the coagulation index due to platelet aggregation inhibition.
Thus, the presented clinical examples vividly demonstrate that based solely on coagulogram parameters, a physician cannot adequately assess and perform effective hemostasis correction. Ignoring this fact can lead to the rapid development of life-threatening complications for the patient. Evaluation of the clotting system and monitoring of antiplatelet and anticoagulant therapies should be based solely on a comprehensive analysis. Analysis of the hemostatic system, along with coagulogram data, must necessarily include TEG parameters, which are crucial. Additionally, other clinical and laboratory indicators, as well as immunological and biochemical research data, should be taken into account. These indicators characterize the state of the body's internal environment and directly or indirectly influence hemostasis. For example, this includes liver enzyme activity, platelet count, homocysteine level, immunoglobulins, acute-phase proteins, blood oxygen levels, and so on.
About D-Dimer Levels
Recently, especially during the COVID-19 period, there has been widespread use of D-dimer level analysis among clinicians. Based on D-dimer levels, clinicians attempt to assess the effectiveness of anticoagulant therapy. However, it is important to clarify certain key aspects of this study and provide clarity on the interpretation of the obtained data. Firstly, it should be noted that D-dimers are products of the breakdown of fully formed blood clots, which are formed as a result of fibrinolysis [49]. Moreover, the key word here is "breakdown." Elevated levels of D-dimers appear only when a large number of fully formed blood clots, resulting from fibrinolysis activation, start to rapidly break down rather than form. This means that during acute thrombosis (such as in pulmonary embolism, myocardial infarction, stroke, etc.), for a certain period of time ranging from several tens of minutes to hours, D-dimer levels may remain within the normal range, and the anticoagulant therapy conducted according to protocol may be considered adequate. As the D-dimer levels increase, often in the absence of fibrinolysis activity data, clinicians mistakenly believe that it is necessary to increase the dosage of anticoagulants. In doing so, they overlook the fact that D-dimers inhibit the polymerization process of fibrin monomer complexes, and their rapid increase can lead to a transition from hypercoagulation to hypocoagulation and the development of bleeding [50]. Blindly following protocols and instructions, doctors fail to consider the fact that many pharmacological agents can also influence fibrinolysis activity to varying degrees.
It is also important to emphasize that in cases of thrombosis, the platelet count does not correlate with their functional activity. Therefore, thrombocytosis or thrombocytopenia is not absolute indications or contraindications for the use of pharmacological hemostatic agents (not to be confused with platelet transfusion!).
FINDINGS
Proper correction of the hemostatic system can only be carried out by a highly qualified specialist - a clinical transfusiologist, who possesses a comprehensive understanding of the functioning of the coagulation and anticoagulation systems, platelet and fibrinolysis systems, knows the points of application and mechanisms of action of the pharmacological agents and blood components used, and takes into account the factors of the internal and external environment that influence hemostasis.
CONCLUSION AND SIGNIFICANCE
Empirical administration of anticoagulant and antiplatelet therapies, based solely on instructions and clinical protocols without considering objective analysis of the state of the hemostatic components, is incorrect, ineffective, and most importantly, dangerous for the patient's life. Coagulation profile parameters cannot serve as independent markers for monitoring the hemostatic system. Evaluation of the hemostatic system should only be done based on a comprehensive analysis of coagulation profile data, complete blood count, thromboelastography (TEG), and data from all relevant systems involved.
REFERENCES
- Centers for Disease Control and Prevention. (2020). Coronavirus disease 2019 (COVID-19) in the U.S. Available at: https://www.cdc.gov/coronavirus/2019-ncov/cases-in-us.html
- National Institute of Allergy and Infectious Diseases. (2020). Coronaviruses. Available at: https://www.niaid.nih.gov/diseases-conditions/coronaviruses
- Centers for Disease Control and Prevention. (2020). Interim clinical guidance for management of patients with confirmed coronavirus disease (COVID-19). Available at: https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-guidancemanagementpatients.html
- Nasonov EL. (2020). Koronavirusnaia bolezn' 2019 (COVID-19): razmyshleniia revmatologa. Nauchno-prakticheskaia revmatologiia. 58 (2):123-132.
- Nasonov EL, Beketova TV, Reshetniak TM, et al. (2020). Koronavirusnaia bolezn' 2019 (COVID-19) i immunovospalitel'nye revmaticheskie zabolevaniia na perekrestke problem trombovospaleniia i autoimmuniteta. Nauchno-prakticheskaia revmatologiia. 58(4):353-367.
- Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. (2020). High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 46(6):1089-1098.
- Beun R, Kusadasi N, Sikma M, Westerink J, Huisman A. (2020). Thromboembolic events and apparent heparin resistance in patients infected with SARS-CoV-2. Int J Lab Hematol. 42(Suppl 1):19-20.
- Poissy J, Goutay J, Caplan M, Parmentier E, Duburcq T, Lassalle F, et al. (2020). Pulmonary Embolism in Patients With COVID-19: Awareness of an Increased Prevalence. Circulation. 142(2):184-186.
- Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al. (2020). Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 191:145-147.
- Roncon L, Zuin M, Barco S, Valerio L, Zuliani G, Zonzin P, et al. (2020). Incidence of acute pulmonary embolism in COVID-19 patients: Systematic review and meta-analysis. Eur J Intern Med. 82:29-37.
- Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, Driggin E, et al. (2020). COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J Am Coll Cardiol. 75(23):2950-2973.
- Han H, Yang L, Liu R, Liu F, Wu KL, Li J, et al. (2020). Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med. 58(7):1116-1120.
- Oudkerk M, Kuijpers D, Oudkerk SF, van Beek EJ, et al. (2020). The vascular nature of COVID-19. Br J Radiol. 93(1113):20200718.
- Sakr Y, Giovini M, Leone M, Pizzilli G, Kortgen A, Bauer M, et al. (2020). Pulmonary embolism in patients with coronavirus disease-2019 (COVID-19) pneumonia: a narrative review. Ann Intensive Care. 10:124.
- Zhai Z, Li C, Chen Y, Gerotziafas G, Zhang Z, Wan J, et al. (2020). Prevention and Treatment of Venous Thromboembolism Associated with Coronavirus Disease 2019 Infection: A Consensus Statement before Guidelines. Thromb Haemost. 120(6):937-948.
- Bagot CN, Arya R. (2008). Virchow and his triad: a question of attribution. Br J Haematol. 143(2):180-190.
- Hajjar LA, Costa IBSDS, Rizk SI, Biselli B, Gomes BR, Bittar CS, et al. (2021). Intensive care management of patients with COVID-19: a practical approach. Ann Intensive Care. 11(1):36.
- Baram A, Kakamad FH, Abdullah HM, Mohammed-Saeed DH, Hussein DA, Mohammed SH, et al. (2020). Large vessel thrombosis in patient with COVID-19, a case series. Ann Med Surg (Lond). 60:526-530.
- Morawitz P. (1905). Die Chemie der Blugerinnung. Ergebnisse der Physiologie. 4:307-423.
- Boulton F. (2006). A hundred years of cascading-started by Paul Morawitz (1879–1936), a pioneer of haemostasis and of transfusion. Transfus Med. 16(1):1-10.
- Kang N, Sobieraj DM. (2014). Indirect treatment comparison of new oral anticoagulants for the treatment of acute venous thromboembolism. Thromb Res. 133(6):1145-1151.
- Fred HL. (2017). Skin Necrosis Induced by Coumarin Congeners. Tex Heart Inst J. 44(4):233-236.
- Gray E, Hogwood J, Mulloy B. (2012). The anticoagulant and antithrombotic mechanisms of heparin. Handb Exp Pharmacol. (207):43-61.
- Atkinson HM, Mewhort-Buist TA, Berry LR, Chan AK. (2009). Anticoagulant mechanisms of covalent antithrombin-heparin investigated by thrombelastography. Comparison with unfractionated heparin and low-molecular-weight heparin. Thromb Haemost. 102(1):62-68.
- Găman AM, Găman GD. (2014). Deficiency Of Antithrombin III (AT III)-Case Report and Review of the Literature. Curr Health Sci J. 40(2):141-143.
- Hogan M, Berger JS. (2020). Heparin-induced thrombocytopenia (HIT): Review of incidence, diagnosis, and management. Vasc Med. 25(2):160-173.
- Jin Y, Ji W, Yang H, Chen S, Zhang W, Duan G. (2020). Endothelial activation and dysfunction in COVID-19: from basic mechanisms to potential therapeutic approaches. Signal Transduct Target Ther. 5(1):293.
- Burch M, Cooper B. (2012). Fondaparinux-associated heparin-induced thrombocytopenia. Proc (Bayl Univ Med Cent). 25(1):13-15.
- Walenga JM, Fareed J, Jeske WP, Bıck RL, Samama MM. (2002). Development of a Synthetic Heparin Pentasaccharide: Fondaparinux. Turk J Haematol. 19(2):137-150.
- Walenga JM, Jeske WP, Samama MM, Frapaise FX, Bick RL, Fareed J. (2002). Fondaparinux: A synthetic heparin pentasaccharide as a new antithrombotic agent. Expert Opin Invest Drugs. 11(3):397-407.
- Esmon CT. (1989). The roles of protein C and thrombomodulin in the regulation of blood coagulation. J Biol Chem. 264(9):4743-4746.
- Zhang P, Zhang J, Sun G, Gao X, Wang H, Yan W, et al. (2014). Risk of Budd-Chiari syndrome associated with factor V Leiden and G20210A prothrombin mutation: a meta-analysis. PLoS One. 9(4):e95719.
- Tripodi A. (2013). Results expression for tests used to measure the anticoagulant effect of new oral anticoagulants. Thromb J. 11:9.
- Arnett DK, Blumenthal RS, Albert MA, et al. (2019). 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease. J Am Coll Cardiol. 74(2019):e177-e232.
- Vane JR, Botting RM. (2003). The mechanism of action of aspirin. Thromb Res. 110(5-6):255-258.
- Smith JP, Haddad EV, Taylor MB, Oram D, Blakemore D, Chen Q, et al. (2012). Suboptimal Inhibition of Platelet Cyclooxygenase-1 by Aspirin in Metabolic Syndrome. Hypertension. 59(3):719-725.
- Ruan CH, So SP, Ruan KH. (2011). Inducible COX-2 dominates over COX-1 in prostacyclin biosynthesis: Mechanisms of COX-2 inhibitor risk to heart disease. Life Sci. 88(1-2):24-30.
- Dannhardt G, Kiefer W. (2001). Cyclooxygenase inhibitors-currunt status and future prospects. Eur J Med Chem. 36(2):109-126.
- Khan SU, Lone AN, Kleiman NS, Arshad A, Jain V, Al Rifai M, et al. (2023). Aspirin With or Without Statin in Individuals Without Atherosclerotic Cardiovascular Disease Across Risk Categories. JACC Adv. 2(2):100197.
- Kirkner RM. (2023). Meta-Analysis Throws More Shade Aspirin's Way. Available at: https://www.medscape.com/viewarticle/988326#vp_1
- Obata T, Nagakura T, Masaki T, Maekawa K, Yamashita K. (1999). Eicosapentaenoic acid inhibits prostaglandin D2 generation by inhibiting cyclo-oxygenase-2 in cultured human mast cells. Clin Exp Allergy. 29(8):1129-1135.
- Araujo P, Belghit I, Aarsæther N, Espe M, Lucena E, Holen E. (2019). The Effect of Omega-3 and Omega-6 Polyunsaturated Fatty Acids on the Production of Cyclooxygenase and Lipoxygenase Metabolites by Human Umbilical Vein Endothelial Cells. Nutrients. 11(5):966.
- Simopoulos AP. (2002). The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed Pharmacother. 56(8):365-379.
- Gharami K, Das M, Das S. (2015). Essential role of docosahexaenoic acid towards development of a smarter brain. Neurochem Int. 89:51-62.
- Bos DJ, van Montfort SJ, Oranje B, Durston S, Smeets PA. (2016). Effects of omega-3 polyunsaturated fatty acids on human brain morphology and function: What is the evidence? Eur Neuropsychopharmacol. 26(3):546-561.
- Scorletti E, Byrne CD. (2013). Omega-3 fatty acids, hepatic lipid metabolism, and nonalcoholic fatty liver disease. Annu Rev Nutr. 33:231-248.
- Tobin D, Brevik-Andersen M, Qin Y, Innes JK, Calder PC. (2018). Evaluation of a High Concentrate Omega-3 for Correcting the Omega-3 Fatty Acid Nutritional Deficiency in Non-Alcoholic Fatty Liver Disease (CONDIN). Nutrients. 10(8):1126.
- Quarterman C, Shaw M, Johnson I, Agarwal S. (2014). Intra-and inter-centre standardisation of thromboelastography (TEG®). Anaesthesia. 69(8):883-890.
- Lichey J, Reschofski I, Dissmann T, Priesnitz M, Hoffmann M, Lode H. (1991). Fibrin degradation product D-dimer in the diagnosis of pulmonary embolism. Klin Wochenschr. 69(12):522-526.
- Weisel JW, Litvinov RI. (2013). Mechanisms of fibrin polymerization and clinical implications. Blood. 121(10):1712-1719.