Previous Issues Volume 3, Issue 1 - 2018
Report of the Second Patient with a Homozygous FVII Padua (Arg304GIn) Defect in Northern Argentina
Antonio Girolami1*, Emanuel Sueldo2, Silvia Ferrari1, Gabriela Guerrero2, Elisabetta Cosi1, Mirta Arias2
1University of Padua Medical School, Department of Medicine, Italy.
2Universidad de Buenos Aires, Departamento de Bioquimica Clinica y Hematologia, Italy
Corresponding Author: Antonio Girolami, University of Padua Medical School, Department of Medicine Italy, Tel: 00390 49821 3026; Email: [email protected]
Received Date: 05 Jan 2018 Accepted Date: 19 Feb 2018 Published Date: 22 Feb 2018
Copyright © 2018 Girolami A
Citation: Girolami A, Sueldo E, Ferrari S, Guerrero G, et al. (2018). Report of the Second Patient with a Homozygous FVII PADUA (Arg304Gln) Defect in Northern Argentina. Mathews J Case Rep 3(1): 036.
KEYWORDS
Factor VII; Arg304Gln Mutation; Coagulation Deficiency.
INTRODUCTION
FVII Padua is due to an Arg304Gln mutation in Exon 8. The defect was first described in Italy in 1978-1993 [1, 2].
Subsequently, the defect was seen to be concentrated in a few special areas of the world, namely the Mediterranean countries, Japan, Iran and in the USA among the AfricanAmerican population [3].
Recently, a homozygous case with this defect has been reported from Argentina [4]. Previously only a few heterozygotes were reported from Latin America in Venezuela, Costa Rica and Brazil [5, 6].
We now report another family from Argentina. The propositus is a 79 year old male with an Argentinean background and whose parents were not consanguineous. The patient is not related to the previous family with this defect described in Argentina [4]. The patient is asymptomatic but refers that he showed some excessive bleeding after tooth extractions. At the age of 76, he underwent prostatectomy. Because of the prolonged PT and low FVII he was treated with 40 mcg of activated FVII. No excessive bleeding or any other complication occurred.
The patient has a low FVII level (3% of normal) using a rabbit brain thromboplastin and a near normal level if a human tissue or human recombinant thromboplastins are used in the assay system (60% of normal); FVII activity is 100% of normal using an OX brain thromboplastin. FVII antigen is also normal. On the basis of these clotting discrepancies suggestive of a FVII Padua defect, molecular biology tests were indicated. These were carried out as follows. Nucleic acids were extracted from dried thick drops of whole blood blotted on Whatman paper. For this purpose we used the kit (QiAmp DNA minikit) supplied by QIAGEN Laboratories (Qiagen s.r.l., Milan, Italy).
Amplification of exons 1 to 9 and respective spice junctions of the FVII gene were performed using oligonucleotide primers kindly supplied by Dr. James Harry (University of Texas at Tyler, Tx, U.S.A.) or acquired from Invitrogen (Carlsbad, Ca, U.S.A.).
Primers are listed in Table 1 together with respective annealing temperatures. To investigate Exon 8, two pairs of primers (8AF, 8AR, 8BF, 8BR) were used due to the large size of the Exon 8 (2269 bp). PCR was carried out in a total volume of 15μL with 50ng of genomic DNA, 10mM of each primer, and 9 µl of 2X PCR Master Mix, (Promega, Madison, Wisconsin, U.S.A.). Thermocycling conditions are the following: after an initial denaturation step at 95°C for 5 minutes, amplification was performed for 35 cycles of denaturation at 95°C for 1 minute, respective annealing temperature for 1 minute, extension at 72°C for 2 minutes and final extension at 72° for 7 minutes.
Table 1: Primers list and respective annealing temperature.
| Primer Forward | Primer Reverse | T annealing in °C |
|---|---|---|
| 1F- AGGCTCTCTTCAAATATTTACATC | 1R-CGGGCTGGCTCCTGGATTT | 59° |
| 2F- CCCTAGCTCACAGCATGGCC | 2R-AGGGGAAGGAGGTGATGTTG | 64° |
| 3F- AAGGATGGGCGAACGGGGTG | 3R- TTCACCGCCGCCGTGCAGTG | 60° |
| 4F-GTGAAGGTGCATCTCACGAG | 4R-CCTCTGCAAATATGGGACCC | 63° |
| 5F-GCAGAACACCACTGCTGACC | 5R-AGTGGGACAGGGACTGGTGT | 66° |
| 6F-GCATCTTTCTGACTTTTGTT | 6R-TAGACCCTCAGTGAGTGTC | 57° |
| 7F-AATGTGACTTCCACACCTCC | 7R-GATGTCTGTCTGTCTGTGGA | 62° |
| 8F-GAGGTGGCAGGTGGTGGAAA | 8R-CGGCACAGAACATGTACTCC | 63° |
| 9F-TGATGACCCAGGACTGCCT | 9R-GGGATTTGGTGCCAGGACA | 65° |
After the amplification step, we passed directly to the sequencing, without the gel detection, because the PCR protocol for this gene has been optimized (2). PCR products were bidirectionally sequenced using the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit and ABI3130 Genetic Analyzer (Applied Biosystems, Foster City, Ca, U.S.A.).
The patient was found indeed to be homozygous for the Arg304Gln mutation (Figure.1). The children of the propositus were found to be heterozygotes for the same mutation. Only one of them, a 47 year old male, had noted excessive bleeding after a tooth extraction. The wife of the propositus was instead normal.
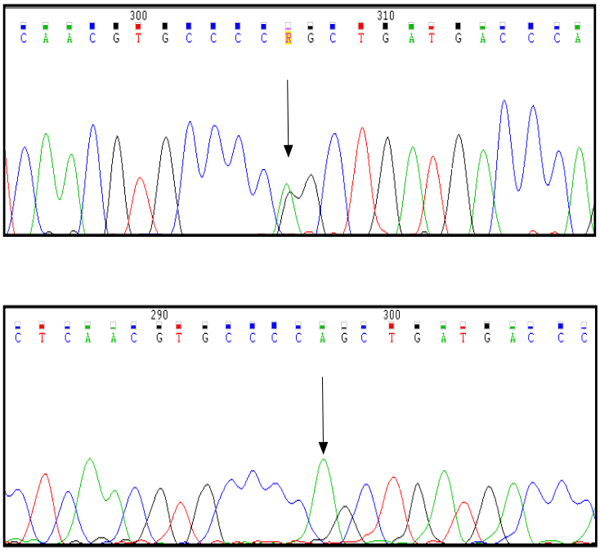
Sequencing results in a heterozygous daughter of the propositus (A) and in the propositus who is a homozygote (B). No other genetic abnormalities were present in the two patients.
The discovery of two unrelated homozygous patients in such a short period of time in the same country suggests that other similar cases may exist. Due to the heavy emigration to Argentina from Southern Europe or Mediterranean Countries, that occurred in the past, the finding is not surprising.
The observation of these two cases indicates that Argentina may represent another country or area in which the defect is frequent.
A few other cases of FVII deficiency have been described in Argentina but there is no molecular biology confirmation that could define the type of defect and their having the FVII Padua mutation [7, 8]. The present observation has once again underscored the diagnostic value of the discrepancies seen in FVII assays using rabbit brain, human placenta, human recombinant and ox-brain derived thromboplastins [9].
Argentina should be added to the list of areas or countries where FVII Padua is relatively common, namely the Mediterranean area, Japan and USA [3]. However, further studies like FVII mutation screening in a large number of patients is needed to establish this observation.
REFERENCES
- Girolami A, Fabris F, Zanon R, Ghiotto G, et al. (1978). Factor VII Padua: a congenital coagulation disorder due to an abnormal factor VII with a peculiar activation pattern. J Lab Clin Med. 91(3): 387-395.
- James HL, Kumar A, Girolami A, Hubbard J, et al. (1991). Variant coagulation factors X and VII with point mutations in highly conserved motif in the substrate binding pocket. Comparative molecular modelling. Thromb Haemost. Abstract book. 65(Suppl.): 937.
- Girolami A, Berti de Marinis G, Bonamigo E and Allemand E. (2010). Worldwide diffusion of FVII Arg304Gln coagulation defect (FVII Padua). Eur. J. Haematol. 86(2): 135-139.
- Girolami A, Arias M, Sueldo E, Scoles G, et al. (2016). First report of homozygous factor VII Padua (Arg304Gln) defect in a family from Argentina. Hemat. Med.Onc. 1(1): 1-5.
- Herrmann FH, Wulff K, Auerswald G, Schulman S, et al. (2009). Greifswald Factor FVII Deficiency Study Group. Factor VII deficiency: clinical manifestation of 717 subjects from Europe and Latin America with mutations in the factor 7 gene. Haemophilia. 15(1): 267-280.
- Rodrigues DN, Siqueira LH, Galizoni AM, Arruda VR, et al. (2003). Prevalence of factor VII deficiency and molecular characterization of the F7 gene in Brazilian patients. Blood Coagul Fibrinolysis. 14(3): 289-292.
- Bergna LJ, Dours MT, Gonzalez de MN, Martinez AA. Deficiencia congenita de factor VII.Presentacion de 4 casos. Medicina. 41(Suppl): 242-248.
- Saavedra H, Olerto D, Serrano J, Miranda Soto R, et al. (2013). Deficit de FVII hereditario. Hematologia. 17(Suppl.): 176.
- Girolami A, Berti de Marinis G, Bonamigo E, Sartori R, et al. (2010). Ox brain versus rabbit brain thromboplastin assays are the best tool for a preliminary diagnosis of the Arg304Gln factor VII defect (FVII Padua). Acta Haematol. 124(4): 229-234.