Information Links
Related Conferences
Previous Issues Volume 7, Issue 3 - 2022
Aplastic Anemia in Wilson’s disease: A Rare Complication
Aritra Saha1,*, Jinku Ozah1, Mriganka Deka1, Ajit Kumar Pegu2, Somnath Saha Roy1, Sofiur Rahman1, Yash Duseja1
1Postgraduate trainee, Department of Medicine, Assam Medical College & Hospital, Dibrugarh, Assam, India
2Professor & Head, Department of Medicine, Assam Medical College & Hospital, Dibrugarh, Assam, India
*Corresponding author: Dr Aritra Saha, Department of Medicine, Assam Medical College & Hospital, Dibrugarh, PIN 786002, Assam, India. Tel: +91-9401650438, Email: [email protected]
Received Date: December 9, 2022
Publication Date: December 15, 2022
Citation: Saha A, et al. (2022). Aplastic Anemia in Wilson’s Disease: A Rare Complication. Mathews J Case Rep. 7(3):78.
Copyright: Saha A, et al. (© (2022)
ABSTRACT
Wilson’s Disease (WD) is an autosomal recessive disease characterized by an excess of copper buildup in the body due to mutations in ATP-7B gene. Wilson’s disease primarily affects liver and basal ganglion of brain, and therefore patients predominantly have hepatic, neuro-psychiatric manifestations, which may vary from an asymptomatic state to life threatening fulminant hepatic failure. Diagnosis depends on a high clinical suspicion, typical neurological symptoms, presence of KF ring, decreased serum ceruloplasmin concentration, raised 24 hours urinary copper levels and genetic studies in selected individuals.
Pancytopenia due to bone marrow failure is an extremely rare finding in a patient with WD and the exact mechanism for the same is unknown. Here we present the case of a 22-year-old female who presented with features of severe anemia and was eventually diagnosed as a case of WD with bone marrow failure and to the best of our knowledge is a third such case reported worldwide.
Keywords: Wilson’s disease, Aplastic anemia, Bone marrow failure
INTRODUCTION
Wilson’s disease is an autosomal recessive inherited disorder characterized by impaired biliary copper excretion that results in the accumulation of copper in various organs including the liver, the cornea, the brain, etc. The gene of Wilson’s disease lies on chromosome 13 and codes for a copper transporting p-type ATPase -ATP 7B and mutation in the ATP-7B gene causes failure in excretion of copper from hepatocyte into bile and a defective synthesis of ceruloplasmin [1]. Ceruloplasmin, a serum glycoprotein, is the predominant copper carrier in blood and patients with Wilson’s disease have low levels of ceruloplasmin [2]. The global prevalence of Wilson’s disease is 12.7 per 100000 [3].
Wilson’s disease has a heterogenous clinical manifestation, with predominantly hepatic and neuropsychiatric involvement, however many patients are asymptomatic [4]. Hepatic manifestations are seen in the first decade of life and vary from being asymptomatic to a stage of fulminant liver failure [5]. Among neurological manifestations, extrapyramidal features like Parkinsonism, dystonia and ataxia are very common [6]. Craniofacial dystonia may cause typical “Wilsonian Facies” characterized by dull looking face with vacuous smile and drooling of saliva [5]. While hepatic and neurological manifestations are the commonly encountered ones in WD, there may be involvement of other organs as well, such as:
i. Cardiac: cardiomyopathy, cardiac death and arrythmias [7].
ii. Skeletal: Rickets, osteomalacia and osteoporosis [8,9].
iii. Renal: Nephrolithiasis and renal tubular acidosis [10].
iv. Endocrine: Hypogonadotropic hypogonadism, diabetes, growth retardation, hyperparathyroidism, repeated miscarriages and infertility [9].
v. Others features include skin changes, immunological dysfunction, lipoma etc. [8]
Diagnosis of WD is dependent upon both clinical and laboratory features of altered copper metabolism [11]. Usually, demonstration of KF ring along with low serum ceruloplasmin (<0.1g/L) is sufficient to diagnose WD and the commonly utilized investigations include serum ceruloplasmin, 24-hours urinary copper, serum free copper, hepatic copper load and slit lamp test to detect KF ring [12]. A diagnostic scoring system was proposed during the 8th International Meeting on Wilson’s Disease, in Leipzig in 2001, which includes:
a) Clinical parameters: KF ring and Neurological symptoms.
b) Laboratory parameters: Serum ceruloplasmin, direct antibody/Coomb’s test, hepatic copper and urinary copper.
c) Mutation analysis.
A score of more than 3 establishes diagnosis, whereas the diagnosis is very unlikely with a score of less than 3 [12,13].
The main treatment of WD revolves around the use of D-Penicillamine, trientine, zinc, tetrathiomolybdate and dimercapol [12] but the treatment of choice for WD patients presenting with acute liver failure is liver transplantation [14]. There is also a potential role of antioxidants, such as vitamin E in WD [15]. Apart from the drugs affecting copper metabolism, other drugs are also used for symptomatic relief, for example, beta blockers for essential tremor, trihexyphenidyl, baclofen and clonazepam for dystonic tremors, levodopa and amantadine for Parkinson’s disease and botulinum toxin for drooling, etc. [16]
Hematological manifestations are relatively less common. Leukopenia, thrombocytopenia and hemolytic anemia due to Wilson’s disease have been reported but pancytopenia without hypersplenism is extremely rare and there has been only a couple of reports for the same [17-19].
Here, we report the case of a 22-year-old female who presented to us with the features of severe anemia and further investigations revealed pancytopenia. On further evaluation, she was diagnosed as Wilson’s disease with aplastic anemia.
CASE PRESENTATION
A 22-years-old female, presented to our institute with a 15 days history of generalized weakness and easy fatigability, without any history suggestive of upper abdominal discomfort, passage of dark color urine, menorrhagia, tingling sensation in her limbs, ulceration around her mouth, past history of jaundice or repeated blood transfusion. There was no history suggestive of any connective tissue disorder. There was no history of similar illness in the past or in her family. No history of prior treatment with any allopathic or herbal medications.
Clinical examination was unremarkable except for marked pallor, mild icterus and a palpable liver without any palpable lymph nodes. Other organs were not palpable. There were also no signs suggestive of skeletal anomaly or skin changes. Investigations were planned accordingly to evaluate the causes of anemia, especially hemolytic anemia, owing to the presence of icterus and hepatomegaly.
Investigations revealed a picture of pancytopenia, with a total leucocyte count of 2700/cumm, Hemoglobin 6.1 g/dl, corrected reticulocyte count of 0.2% and a platelet count of 60,000/cumm. Other parameters were essentially normal, except for total bilirubin, which was slightly elevated, with predominantly unconjugated hyperbilirubinemia (Table 1). Patient tested negative for the viral infections known to causes pancytopenia, such as hepatitis B, hepatitis C, HIV, EBV, CMV and ParvoB [19]. Autoimmune profile was unremarkable and included a negative ANA and DCT, with a normal serum IgG level. Patient’s hemoglobin electrophoresis was normal.
Bone marrow aspiration was suggestive of hypoplastic marrow and was confirmed by biopsy (Figure 1).
Ultrasound of the abdomen showed enlarged liver without splenomegaly or any genitourinary tract abnormalities and doppler study confirmed normal portal flow, with a portal vein diameter of 0.9 cm.
Owing to the enlarged liver, we performed a slit lamp examination, where we found a KF ring (Figure 2). Eventually, we tested for serum ceruloplasmin, which was low (10mg/dl) and a 24-hour urinary copper, which was high (324 mcg) and therefore the diagnosis of Wilson’s disease was made.
As the patient was mainly symptomatic because of the bone marrow failure, without any evidence of neurological involvement, we started the patient on Tab Zinc acetate 50 mg twice daily with regular transfusion of blood products. Copper chelator, such as D-Penicillamine was not used in this case because it is known to cause cytopenia. Patient is currently under.
Table 1: Laboratory reports.
|
Investigations |
Value |
Interpretation |
|
Hemoglobin (mg/dl) |
5.7 |
Low |
|
Reticulocyte count |
0.3 |
Low |
|
Total leucocyte count (per cumm) |
1600 |
Low |
|
Platelet count (per cumm) |
90,000 |
Low |
|
Total Bilirubin (mg/dl) |
2.93 |
High |
|
Indirect Bilirubin (mg/dl) |
2.13 |
High |
|
AST |
51 |
Marginally elevated |
|
ALT |
47 |
Marginally elevated |
|
A/G |
0,74 |
|
|
LDH |
220 |
Normal |
|
Serum Ferritin |
>1000 mg/ml |
High |
|
Direct Coomb’s Test |
Negative |
|
|
ANA |
Negative |
|
|
Serum IgG (mg/dl) |
937 |
Normal |
|
Bone marrow Biopsy |
Markedly hypocellular |
Abnormal |
|
Serum ceruloplasmin |
10 |
Low |
|
24 hours urinary copper |
324 |
High |
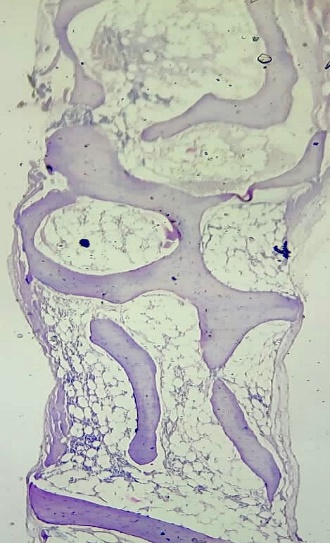
Figure 1: Bone marrow biopsy: Markedly hypocellular marrow with hemopoietic cells replaced by fat.
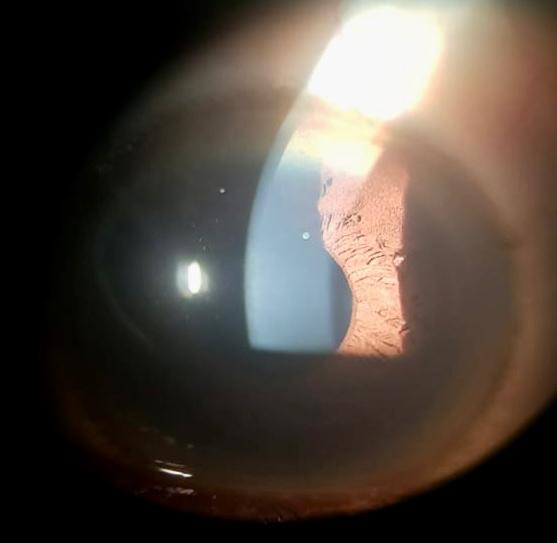
Figure 2: Slit Lamp Examination: Keyser Fleischer Ring.
DISCUSSION
Wilson’s disease is an autosomal recessive disorder characterized by excessive copper accumulation in various organs, especially in the liver, brain and cornea, due to genetic mutations in ATP7B gene on chromosome number 13 [20]. The disease has a prevalence of 1 in 30,000 individuals worldwide and is mainly seen in the age group of 4 to 40 years [21,22]. The common clinical manifestations are due to the involvement of the liver and brain, such as jaundice, abdominal pain, personality changes, dystonia, etc. [5,23]. Kayser-Fleischer ring (KF ring) is an ophthalmic sign, that occurs as a result of excessive copper deposition in the Descemet membrane and is a common finding in Wilson’s disease, however it may be absent in up to 50% patients with only hepatic involvement or patients who are in the pre-symptomatic stage. KF ring has also been described in other conditions such as primary biliary cirrhosis [24,25].
The fundamental diagnostic elements for WD include serum ceruloplasmin, 24 hours urinary copper levels, serum free copper, dry weight of copper in liver, presence of KF ring and ATP-7B mutation study [26].
Chelating agents such as D-Penicillamine, Trientine, Zinc acetate, Dimercaptosuccinate, etc. are the mainstay of treatment. Surgical intervention is required in patients not responding to conservative management, TIPS (transjugular intrahepatic portosystemic shunt) and liver transplantation are couple of them [27]. Liver transplantation is the treatment of choice in patients of WD presenting with acute liver failure [14].
Among the hematological manifestations, hemolytic anemia is seen in 5 to 10 % of the individuals [4,11,18], however leucopenia and thrombocytopenia have been also reported [19]. Hypersplenism is characterized by pancytopenia and therefore is a common finding in patients with cirrhosis [28]. Pancytopenia in patients with Wilson’s disease, without underlying cirrhosis and portal hypertension is relatively rare. Aplastic anemia is characterized by bone marrow failure due to pathology in the bone marrow precursor cells, resulting in a cellularity of less than 25% and tri-lineage depletion [29].
There are only a couple of case reports of aplastic anemia in Wilson’s disease worldwide. Sharma et al. reported aplastic anemia in an 8 year old female with Wilson’s disease in 2012 [18]. In 2015, Acipayam et al. reported a similar case in an 11-year-old female in Turkey [17]. However, there are reports of cytopenia due to the use of copper chelator (penicillamine) in patients with WD and therefore we avoided it in our patient [30,31].
Bone marrow failure due to copper deficiency has been documented in case reports and series [32,33], but the exact pathophysiology of bone marrow failure in a copper excess state such as in WD is not well understood and further research is required in this field for better understanding and management of such cases.
To the best of our knowledge, this is probably the third case of WD in the world to present with aplastic anemia as the initial feature.
CONCLUSION
Aplastic anemia is an underreported complication of Wilson’s disease. The cause of bone marrow failure in WD is not well understood and warrants further studies in this field.
CONFLICTS OF INTEREST
None
FUNDING
Not applicable.
REFERENCES
- Ferenci P. (1998). WILSON’S DISEASE. Clin Liver Dis. 2(1):31-49.
- Terada K, Kawarada Y, Miura N, Yasui O, Koyama K, Sugiyama T. (1995). Copper incorporation into ceruloplasmin in rat livers. Biochim Biophys Acta BBA - Mol Basis Dis. 1270(1):58-62.
- Gao J, Brackley S, Mann JP. (2019). The global prevalence of Wilson disease from next-generation sequencing data. Genet Med. 21(5):1155-1163.
- Stremmel W, Meyerrose KW, Niederau C, Hefter H, Kreuzpaintner G, Strohmeyer G. (1991). Wilson disease: clinical presentation, treatment, and survival. Ann Intern Med. 115(9):720-726.
- Nagral A, Sarma MS, Matthai J, Kukkle PL, Devarbhavi H, Sinha S, et al. (2019). Wilson’s Disease: Clinical Practice Guidelines of the Indian National Association for Study of the Liver, the Indian Society of Pediatric Gastroenterology, Hepatology and Nutrition, and the Movement Disorders Society of India. J Clin Exp Hepatol. 9(1):74-98.
- Taly AB, Meenakshi-Sundaram S, Sinha S, Swamy HS, Arunodaya GR. (2007). Wilson Disease: Description of 282 Patients Evaluated Over 3 Decades. Medicine (Baltimore). 86(2):112-121.
- Kuan P. (1987). Cardiac Wilson’s disease. Chest. 91(4):579-583.
- Dzieżyc K, Litwin T, Członkowska A. (2017). Other organ involvement and clinical aspects of Wilson disease. Handb Clin Neurol. 142:157-169.
- Kapoor N, Shetty S, Thomas N, Paul TV. (2014). Wilson’s disease: An endocrine revelation. Indian J Endocrinol Metab. 18(6):855-857.
- Zhuang XH, Mo Y, Jiang XY, Chen SM. (2008). Analysis of renal impairment in children with Wilson’s disease. World J Pediatr WJP. 4(2):102-105.
- Gow PJ, Smallwood RA, Angus PW, Smith AL, Wall AJ, Sewell RB. (2000). Diagnosis of Wilson’s disease: an experience over three decades. Gut. 46(3):415-419.
- European Association for Study of Liver. (2012). EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol. 56(3):671-685.
- Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. (2003). Diagnosis and phenotypic classification of Wilson disease. Liver Int Off J Int Assoc Study Liver. 23(3):139-142.
- Catana AM, Medici V. (2012). Liver transplantation for Wilson disease. World J Hepatol. 4(1):5-10.
- Fryer MJ. (2009). Potential of vitamin E as an antioxidant adjunct in Wilson’s disease. 73(6):1029-1030.
- Litwin T, Dušek P, Członkowska A. (2017). Symptomatic treatment of neurologic symptoms in Wilson disease. In: Handbook of Clinical Neurology [Internet]. Elsevier: 211-223.
- Acıpayam C, Altunay A, İlhan N, Atcı N. (2015). Wilson’s Disease Presenting With Pancytopenia. Mustafa Kemal Üniversitesi Tıp Derg. 6.
- Sharma SK, Singh AK, Singh PK, Seth T, Mishra P, Vanathi M, et al. (2012) Aplastic Anemia in a Patient With Wilson’s Disease. J Hematol. 1(4-5):100-101.
- Hogland HC, Goldstein NP. (1978). Hematologic (cytopenic) manifestations of Wilson’s disease (hepatolenticular degeneration). Mayo Clin Proc. 53(8):498-500.
- Deguti MM, Genschel J, Cancado ELR, Barbosa ER, Bochow B, Mucenic M, et al. (2004). Wilson disease: novel mutations in the ATP7B gene and clinical correlation in Brazilian patients. Hum Mutat. 23(4):398.
- Chaudhry HS, Anilkumar AC. (2022). Wilson Disease. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing.
- Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. (2007). Wilson’s disease. Lancet Lond Engl. 369(9559):397-408.
- Hedera P. (2019). Wilson’s disease: A master of disguise. Parkinsonism Relat Disord. 59:140-145.
- Pandey N, John S. (2022) Kayser-Fleischer Ring. In: StatPearls. StatPearls Publishing.
- Schilsky ML. (2018). Kayser-Fleischer Ring - an overview. In: Wilson Disease and Related Disorders. 253-268.
- Walshe JM. (2011). The eye in Wilson disease. QJM Mon J Assoc Physicians. 104(5):451-453.
- GilroyRK, Shah R, Piper MH, Roy PK. (2019) Wilson Disease Treatment & Management. Gastroenterol.
- Lv Y, Lau WY, Li Y, Deng J, Han X, Gong X, et al. (2016). Hypersplenism: History and current status. Exp Ther Med. 12(4):2377-2382.
- Moore CA, Krishnan K. (2022). Aplastic Anemia. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing.
- Wiggelinkhuizen M, Tilanus MEC, Bollen CW, Houwen RHJ. (2009). Systematic review: clinical efficacy of chelator agents and zinc in the initial treatment of Wilson disease. Aliment Pharmacol Ther. 29(9):947-958.
- Rau AR, Usha M, Mallya P, Rau ATK. (2014). Cytopenia and Bone Marrow Dysplasia in a Case of Wilson’s Disease. Indian J Hematol Blood Transfus. 30(Suppl 1):433-436.
- Wazir SM, Ghobrial I. (2017). Copper deficiency, a new triad: anemia, leucopenia, and myeloneuropathy. J Community Hosp Intern Med Perspect. 7(4):265-268.
- Haddad AS, Subbiah V, Lichtin AE, Theil KS, Maciejewski JP. (2008). Hypocupremia and bone marrow failure. Haematologica. 93(1):e1-e5.