Information Links
Related Conferences
Current Issue Volume 11, Issue 2 - 2026
Alternating Hemiplegia of Childhood in a Clinical Context: A Case Presentation
Ernesto Portuondo Barbarrosa1,*, Gretel Huerta Peres2
1First-Degree Specialist in Comprehensive General Medicine, Second Degree Specialist in Pediatrics and Neurology, Professor and Assistant Researcher, Head of Clinical Neurophysiology Department, Pediatric Teaching Hospital Centro Habana, Cuba
2First-Degree Specialist in Comprehensive General Medicine and Clinical Genetics, Master in Genetic Counseling, Assistant Professor, Pediatric Teaching Hospital Centro Habana, Cuba
*Corresponding author: Dr. Ernesto Portuondo Barbarrosa, First-Degree Specialist in Comprehensive General Medicine, Second Degree Specialist in Pediatrics and Neurology, Professor and Assistant Researcher, Head of Clinical Neurophysiology Department, Pediatric Teaching Hospital Centro Habana, Cuba, Phone: 054495401, ORCID: 0000-0001-9578-6496, Emails: [email protected]; [email protected]
Received Date: April 07, 2026
Published Date: April 22, 2026
Citation: Barbarrosa EP, et al. (2026). Alternating Hemiplegia of Childhood in a Clinical Context: A Case Presentation. Mathews J Case Rep. 11(2):221.
Copyrights: Barbarrosa EP, et al. © (2026).
ABSTRACT
Introduction: Alternating hemiplegia of childhood (AHC) is a rare disease characterized by recurrent episodes of alternating hemiplegia or quadriplegia that improve during sleep and vary in duration. Onset occurs before 18 months of age and is preceded by abnormal eye movements, accompanied by tonic and dystonic attacks with subsequent developmental regression. The main etiology is related to the ATP1A3 genetic mutation. Clinical Case: A 16-month-old male preschooler with no significant personal or family history. He had previously experienced psychomotor developmental delay in motor skills and language, accompanied by abnormal eye movements. At 12 months of age, he had two episodes: the first, in the context of an arboviral infection, resulted in diplegia; the second, following a prolonged bus trip, involved left hemiplegia lasting 7 days, with spontaneous recovery and no alteration of consciousness, followed by dystonic episodes. Complementary studies of cerebrospinal fluid cytochemistry and culture, multislice cranial CT scan, and normal metabolic studies in urine and serum were performed. Electroencephalography showed slow delta global activity without paroxysms. Treatment was initiated with flunarizine 5 mg daily at night. Follow-up was in the Neuropediatrics outpatient clinic. Genetic testing was limited. Conclusion: Alternating hemiplegia of childhood (AHC) is an entity whose diagnosis is essentially clinical, and our patient met the clinical criteria. Treatment with flunarizine is important to improve prognosis and quality of life.
Keywords: Alternating Hemiplegia of Childhood, Flunarizine.
INTRODUCTION
Alternating hemiplegia of childhood (AHC) is a rare and uncommon disease. Its incidence is estimated at approximately 1 per 1,000,000 live births, but this number may be underestimated due to the variability of symptoms and clinical signs in different epidemiological studies [1-5]. It was first described by Verret and Stele in 1971 as alternating hemiplegic migraine [6]. In 1980, Krakelog and Aircardi differentiated AHC from other hemiplegias in their publication and established diagnostic criteria that are still valid today, because the paroxysmal and non-paroxysmal symptoms, in conjunction with psychomotor developmental regression (PMD) and cognitive impairment, allowed its exclusion from that hypothesis [7].
The pathophysiological mechanisms of autoimmune hepatitis (AIH) remain unknown and enigmatic. Its genetic etiology, in combination with environmental factors, has recently been clarified. It has been identified as being related to the ATP1A3 gene (previously associated with parkinsonism-dystonia of sudden onset). This gene is located on chromosome 19q13.2 in more than 70% of patients and has an autosomal dominant inheritance pattern, although de novo cases are more common. There are more than 60 phenotypic variants of the disease, the most frequent being D801N (43%), E815K (16%), L839P and G947R (11%), and other variants involving the ATP1A2 gene (1q23.3), CACNA1A, and SLC1A3. It is considered a channelopathy with inhibited activity of the Na + /K + ATPase pump, a subunit that encodes and is expressed exclusively in neurons and cardiac muscle cells; this justifies a favorable clinical response to flunarizine, a non-selective blocker of voltage-gated sodium and calcium channels [1,3,6,8].
Clinically, it is characterized by a sequence of successive episodes of hemiplegia that alternately affect one side of the body, and less frequently quadriplegia or diplegia, which generally begin before 18 months of age, are of variable duration (from minutes, hours, days, or an undefined period), disappear during sleep, and return during wakefulness. These episodes are unaffected by any alteration of consciousness and are accompanied by other earlier-appearing clinical manifestations such as abnormal eye movements (paroxysmal nystagmus, upward eye deviation, and eye rolling), autonomic symptoms, tonic, dystonic, and choreoatetoxic attacks that can be confused with epileptic seizures, and in some patients, these seizures may appear later [1-3,6,9].
The diagnosis of AIH is essentially clinical and relies on diagnostic criteria, but demonstrating a genetic variant is of great value in confirming the diagnosis, estimating the prognosis, and providing genetic counseling [1,6].
The aim of our article is to present a clinical case of onset at 15 months of age with prior delayed puberty, without risk factors. The case meets the clinical criteria, but genetic confirmation cannot be demonstrated due to limitations in the studies.
CLINICAL CASE
A 16-month-old male preschooler, of Cuban nationality and from the province of Santiago de Cuba, rural area. Son of young, non-consanguineous parents, firstborn with no significant personal, prenatal, perinatal and postnatal medical history, with a gestational age of 39.2 weeks, vaginal delivery with APGAR (9/9), Weight/ 3000 gr, height/49cm and head circumference/ 34 cm and a hospital stay of 3 days with no significant family medical history.
Previous developmental delay: In the motor domain, head control at 5 months, rolling and sitting with support at 7 months, did not crawl and achieved standing with support at 12 months, and has attempted to take steps with support since 13 months. In the fine motor domain, the pincer grasp has not yet been acquired, and the child transfers objects from one hand to the other. Regarding language and hearing, the child communicated with cooing until 12 months and a few monosyllables, with otoacoustic emissions at birth and normal auditory evoked potentials. Socially, the child smiles, recognizes mother and family members, and blows kisses. Before 12 months, there were brief, sporadic episodes of upward gaze deviation, with variable frequency, without clinical evaluation.
History prior to 12 months in the context of an Arbovirus epidemic (dengue and chikungunya) in his province, admission with acute febrile syndrome of 5 days of evolution with maculopapular rash on the 3rd day of fever and restlessness and in clinical context "weakness of the lower limbs with immobility" and interpreted as a motor deficit without specifying the cause of duration of 7 days with spontaneous recovery.
At 15 months of age, during a 16-hour bus trip from Santiago de Cuba to Havana to visit relatives, upon arrival home, the child experienced weakness in the left side of the body and instability while standing. This was not related to fever or other infectious symptoms, and there was no evidence of contact with medications or other toxic substances, trauma, or altered mental status. The child was admitted to the Central Havana Teaching Pediatric Hospital through the emergency department and placed in the progressive care unit.
On physical examination and medical evaluation: the child was awake, interacted with the environment, and defended himself during the examination, with no evidence of altered mental status. During the examination of eye movements, mild, fatigable horizontal nystagmus was noted, with no cranial nerve palsies. Left hemiplegia was present with diminished deep tendon reflexes; no clonus or Babinski sign was observed. When seated with support, the child fell to the left side.
Additional tests:
Hematocrit / 0.34 ≈ 11.2 g/L of Hemoglobin, leukogram with leukocytes/7.2, polymorphonuclear cells 0.52, lymphocytes 0.48, normal coagulation panel with platelet count/ 330. Ionogram/blood gas analysis - normal with anion gap (18), transaminases glutamic-oxaloacetic (25 U/L), glutamic-pyruvic (28 U/L), lactate dehydrogenase (222 U/L) within normal parameters. Cerebrospinal fluid (CSF) with clear and transparent appearance, cells 0 x mm3, CSF glucose/ 3 mmol/L with blood glucose 4 mmol/L, proteins / 0.25 g/L within normal parameters with Gram stain and CSF culture without bacterial growth.
Urine metabolic tests (UMT) were performed, with amino acid and organic acid profiles using tandem spectrometry, with normal results. Multislice computed tomography (CT) showed no evidence of structural abnormalities (Figure 1a and 1b); the first electroencephalogram (EEG) during wakefulness and sleep showed slow delta activity (4-4.5 Hz), Figure 2a and 2b. Echocardiogram showed no structural or cardiac function abnormalities.
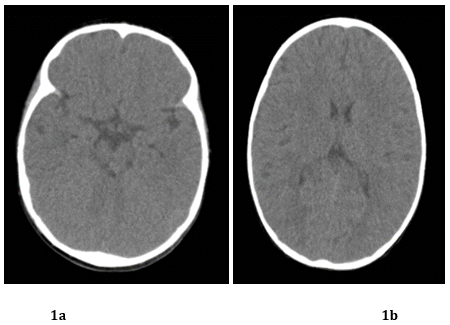
Figure 1a & 1b. Multislice CT scan, axial sequence with no evidence of structural abnormalities and normal ventricular system.
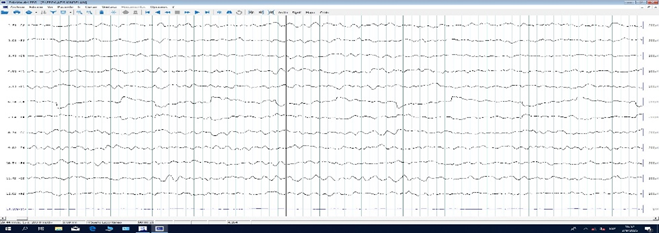
Figure 2a
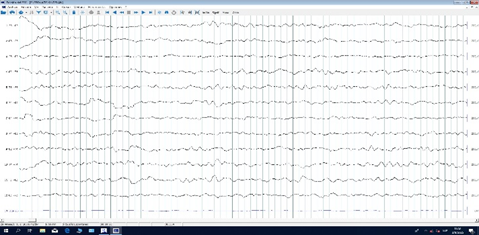
Figure 2b
Figure 2a & 2b. 20-channel EEG, 10-20 system, spontaneous sleep, stage 1-2, with global slow delta activity (4-4.5Hz).
During his clinical evolution with a hospital stay of 15 days, he had spontaneous recovery of symptoms in 7 days and it was subsequently noted that he had brief waking events with opisthotonus posture, with video affiliation provided by the family and without alteration of consciousness; with a 2nd EEG of the same characteristics and without paroxysmal activity and being interpreted as dystonic events.
Based on the clinical presentation and patient history, with normal complementary studies allowing for differential diagnosis with other conditions and meeting clinical criteria, a diagnosis alternating hemiplejia of childhood (AIH) was determined in conjunction with the genetics service. Treatment was initiated with flunarizine 5 mg/24 hours at night. During outpatient follow-up over two months, there was only one episode affecting the right side of the body, lasting 48 hours, with no improvement in motor function. The patient communicated using jargon and monosyllables, stood with support, and took a few steps with support. The patient was integrated into a community.
DISCUSSION
AIH is a rare, uncommon, and low-incidence condition that is difficult for medical personnel to recognize and identify. In early childhood, these episodes are often confused with infant seizures, making epilepsy a differential diagnosis to consider [1,3,10]. Other differential diagnoses would include: Moyamoya angiopathy or other arteriovenous malformations, ischemic stroke, inborn errors of metabolism (organic acidurias, urea cycle disorders, and pyruvate dehydrogenase deficiency), mitochondrial encephalopathies (MELAS and Kearns-Sayre), hypokalemic periodic paralysis, early epileptic encephalopathy with infantile spasms (EEIS) vs. West syndrome, Todd's postictal paralysis, CAPOS syndrome (cerebellar ataxia, areflexia, optic atrophy, and pes cavus), RPD syndrome (rapid onset dystonia-parkinsonism) at a later age that share the same ATP1A3 gene, and familial hemiplegic migraine [9,11-14].
The disease is characterized by clinical manifestations that can be organized into different stages or phases [6-9].
Phase one (first months of life): Abnormal eye movements (nystagmus, rolling, upward gaze deviation) with or without delay in DPM.
Phase two (1 to 5 years): Episodes of alternating hemiplegia while awake, less frequently quadriplegia and diplegia without altered consciousness, of variable duration and frequency; may be accompanied by autonomic symptoms (pallor, flushing, cyanosis, pupillary dilation, vomiting, diarrhea, sweating, or fever). These disappear during sleep and reappear 10 to 20 minutes after waking and are accompanied by abnormal movements (dystonic attacks and choreoathetosis).
Phase 3 (after age 5): Fixed neurological deficits or regression of primary mental status, especially in the cognitive, attention, language, and behavioral domains, with evidence of attention deficit hyperactivity disorder (ADHD), disruptive behavior, and anxiety disorder. Epileptic seizures usually occur after age 6 in approximately 40 to 50% of patients, without a direct relationship to hemiplegia episodes, and present with atonic, focal motor, or generalized tonic-clonic symptoms.
Other manifestations involve cardiorespiratory abnormalities (cardiac conduction disorder) and endocrinopathies of central origin (precocious puberty and growth hormone deficiency) [6,15,16].
Similar to what has been described in the literature, our patient began experiencing symptoms before 12 months of age, with evidence of developmental delays (gross motor skills and language) and abnormal upward gaze deviation. These clinical manifestations precede hemiplegic episodes, as described by Sandoval [1], Guevara-Campos et al. [4], and Egan [13].
The presented plegic events had an onset at 12 months of age; the first was a diplegic episode, and the second was a hemiplegic episode with hyporeflexia lasting approximately 7 days. The second episode was accompanied by dystonic events after the resolution of the motor deficit. It should be noted that both were triggered: the first by fever related to an exanthematous viral infection (suspected arbovirus), and the second after a prolonged bus trip. Several factors are described as triggers for hemiplegic episodes, including infections and fever, prolonged travel, psychological stress, intense exercise, and other environmental factors (bright light, excessive cold or heat, exposure to very cold or hot water), and certain foods such as chocolate [9,17]. These clinical manifestations coincide with other reported clinical cases where most authors describe hemiplegic attacks as not being the initial symptoms of the disease and appearing in the second phase, accompanied by other abnormal movements [1,3,4,6,7,9,13,14].
The diagnosis of AIH is primarily clinical, and the presence or evidence of the genetic alteration or variant is essential to confirm the diagnosis and estimate a prognosis. The clinical criteria were described by Aicardi and Bourgeois [18] and consist of:
a) Start before 18 months of age.
b) Autonomous phenomena.
c) Neurodevelopmental disorders, cognitive and learning difficulties, and/or neurological sequelae such as ataxia, dystonia and choreoathosis.
d) Repeated episodes of hemiplegia that sometimes involve both sides of the body, lasting from minutes to days.
e) Other paroxysmal disorders (abnormal eye movements, diatonic postures).
f) Disappearance of symptoms during sleep and their reappearance upon waking.
g) Exclusion of other disorders that cause recurrent hemiplegia.
Although the criteria proposed an age of onset before 18 months, early studies showed that the age of diagnosis is around 4 years [19]. Typical cases meet criteria a, d, and g [3,6]. In our case, these criteria were met, so we consider it typical.
The genetic studies to be performed include: ATP1A3 gene sequencing (if there is a high clinical probability), multigene panels targeting dystonias, epilepsies, and other abnormal movements, and genome sequencing in cases not confirmed by the previous studies. In our case, due to limitations in genetic studies, the diagnosis is clinical.
Neuroimaging studies such as CT and MRI, especially MRI, are normal in most patients during acute or recurrent episodes of neurological deficits. However, in a group of children with the classic HAI phenotype, diffuse cerebral or cerebellar atrophy and mesial temporal sclerosis are described [6].
The EEG during wakefulness and sleep is normal in most cases at the beginning of the disease and during hemiplegic episodes, without paroxysmal activity or registering slow waking activity for their age, as evidenced by other reported cases [1,3,4,9,14]. This was observed in our patient.
The management and treatment of AIH is complex, considering different aspects: acute episodes, prevention, and comorbidities in its evolution. In the acute phase, benzodiazepines, due to their regulatory action on the GABA system, and other sleep inducers (melatonin, chloral hydrate, diphenhydramine, and phenobarbital) could decrease the duration of attacks due to their sleep-promoting effect, but respiratory distress, desaturations, sedation, or behavioral disturbances would have to be taken into account; with controversial results [6,9].
In preventing seizures, flunarizine is the most commonly used medication and has shown the best results at doses of 5 to 20 mg at night. It has demonstrated efficacy and safety in preventing, reducing the severity and duration (from days to a few hours) of seizures in the long term, with an efficacy rate of approximately 50%. However, it does not alter the prognosis or future course of the condition and is not useful in the treatment of epilepsy or cognitive impairment. Clinical evidence shows that, conversely, patients who did not receive flunarizine experienced severe motor impairment. Other medications used have included topiramate, betamethasone, amantadine, memantine, aripiprazole, oral ATP, coenzyme Q10, acetazolamide, and the ketogenic diet, without demonstrated efficacy. Currently, there are clinical trials underway with cannabidiol [1-4,6,9,10,14]. In our case, we used a dose of 5 mg daily, and in two months of follow-up, the patient only had one episode hemiplejia of short duration. The evolution and prognosis of AIH is uncertain and unfavorable in most affected individuals due to its progressive nature, comorbidities, and the frequency and duration of episodes with motor deficits that do not fully recover in most cases after a crisis. Interdisciplinary follow-up by specialists in neurology, genetics, cardiology, endocrinology, psychiatry, rehabilitation, and speech therapy ensures better.
In our patient, from before the first year, there was a delay in DPM, aggravated after the onset of crises with motor deficit that, in similar case reports [1,2,5,7,17,19], required early intervention in stimulation and rehabilitation.
CONCLUSION
AIH is a rare, enigmatic, and complex disease. Early diagnosis, primarily clinical, requires a thorough medical history, physical examination, and, in some cases, video evidence of episodes with motor deficits. Complementary tests, such as genetic, imaging, and EEG studies, are useful for definitively identifying this condition and establishing a precise differential diagnosis. Early use of flunarizine prevents a worse prognosis by reducing the frequency, severity, and duration of episodes with severe motor deficits, avoiding ineffective treatments, and improving the quality of life for patients and their families.
ACKNOWLEDGEMENTS
None.
CONFLICT OF INTEREST AND FUNDING
There is no conflict of interest and we do not receive external funding.
REFERENCES
- Sandoval F, López F. (2022). Hemiplejía Alternante de la Infancia asociada a variante patogénica del gen ATP1A3 [Alternating Hemiplegia of Childhood associated with a pathogenic variant of the ATP1A3 gene]. Andes Pediatr. 93(1):117-122.
- Neville BG, Ninan M. (2007). The treatment and management of alternating hemiplegia of childhood. Dev Med Child Neurol. 49(10):777-780.
- Ramírez-Zamora M, Ortez-González CI. (2013). Hemiplejía alternante de la infancia. Primer caso clínico descrito en El Salvador [Alternating hemiplegia of childhood. The first clinical case reported in El Salvador]. Rev Neurol. 57(5):212-216.
- Guevara-Campos J, González-de Guevara L, Urbáez-Cano J, Tinedo R, Villamizar M, Rojas L. (2005). Hemiplejía alternante de la infancia tratada como epilepsia. Dos nuevos casos [Alternating hemiplegia of childhood treated as epilepsy. Two new cases]. Rev Neurol. 40(6):351-353.
- Verret S, Steele JC. (1971). Alternating hemiplegia in childhood: a report of eight patients with complicated migraine beginning in infancy. Pediatrics. 47(4):675-680.
- Rengifo MMP, Yepes Madrid N, Valencia Caicedo AM, Gómez Urrego JF. (2025). Unraveling Childhood Alternating Hemiplegia: A scoping review. Acta Neurol Colomb. 41(3):e1911.
- Kräkelog I, Aicardi J. (1980). Alternating hemiplegia in infants: report of five cases. Dev Med Child Neurol. 22(6):784-791.
- Souza AA, Terrey M, George Jr AL, Lutz CM, Liu DR. (2025). In vivo prime editing rescues alternating hemiplegia of childhood in mice. Cell. 188(16):4275-4294.e23.
- Rissardo JP, Vora NM, Singh Y, Kishore S, Caprara ALF. (2024). Navigating the Complexity of Alternating Hemiplegia in Childhood: A Comprehensive Review. Rambam Maimonides Med J. 15(3):e0015.
- Campistol J, Sans- Fitó A, Pineda M, Fernández-Álvarez E. (1990). Alternating hemiplegia in childhood: presentation, evolution and treatment. An Esp Pediatr. 32(4):336-338.
- Shemesh A, Margolin E. (2023). Kearns-Sayre Syndrome. In: Aboubakr S, Ghosh Abu A, Ackley WB, et al., eds. StatPearls. Treasure Island, FL: StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/NBK482341/
- Salles PA, Mata IF, Brünger T, Lal D, Fernandez HH. (2021). ATP1A3-Related Disorders: An Ever-Expanding Clinical Spectrum. Front Neurol. 12:637890.
- Egan RA. (2002). Ocular motor features of alternating hemi-plegia of childhood. J Neuroophthalmol. 22(2):99-101.
- Wei D, Lv K, He J, Xiao B, Long L. (2024). Alternating hemiplegia of childhood misdiagnosed as hysteria: a case report. Acta Epileptol. 6(1):4.
- Pavone P, Pappalardo XG, Ruggieri M, Falsaperla R, Parano E. (2022). Alternating hemiplegia of childhood: a distinct clinical entity and ATP1A3-related disorders: a narrative review. Medicine (Baltimore). 101(31):e29413.
- Wallace K, Greene E, Moya-Mendez M, Freemark M, Prange L, Mikati MA. (2021). Hypothalamic-pituitary dysfunction in alternating hemiplegia of childhood. Eur J Paediatr Neurol. 32:1-7.
- Traut M, Cavagnari BM, Mendez JH, Amartino H. (2012). Hemiplejia alternante de la infancia: presentación de un caso y revisión de la bibliografía. Arch Argent Pediatr. 110(5):86-90.
- Aicardi J, Bourgeois M, Goutieres F. (1995). Alternating Hemiplegia of Childhood: Clinical Findings and Diag-nostic Criteria. In: Andermann F, Aicardi J, Vigevano F (eds). Alternating Hemiplegia of Childhood. New York: Raven Press. pp. 3-18.
- Mikati MA, Kramer U, Zupanc ML, Shanahan RJ. (2000). Alternating hemiplegia of childhood: clinical manifestations and long-term outcome. Pediatr Neurol. 23(2):134-141.