Previous Issues Volume 3, Issue 1 - 2019
The Role of Macrophage Migration Inhibitory Factor (MIF) in Acute Kidney Injury
Jinhong Li, Wenqian Xu, Na Li, Yiqing Zhang, Xiaohua Wang, Chun Tang, Zhihua Zheng*
The Department of Nephrology, Center of Nephrology and Urology, The Seventh Affiliated Hospital, Sun Yat-sen University, China.
Corresponding Author: Zhihua Zheng, The Department of Nephrology, Center of Nephrology and Urology, The Seventh Affiliated Hospital, Sun Yat-sen University, China.
Received Date: Sep 18, 2019 Published Date: Oct 2, 2019 Copyright © 2019 Zheng Z
Citation: Zheng Z. (2019). The Role of Macrophage Migration Inhibitory Factor (MIF) in Acute Kidney Injury. Mathews J Cytol Histol 3(1): 11. ABSTRACT
Macrophage migration inhibitory factor (MIF) is a proinflammatory cytokine implicated in acute and chronic disease, including autoimmune disease, atherogenesis, plaque instability, sepsis, glomerulonephritis, acute kidney injury and CKD. Macrophage migration inhibitory factor (MIF) has emerged as a promising therapeutic target in human diseases including immune disorder, cancer, cardiologic diseases, diabetes, and inflammatory diseases. MIF was also reported contributes to leukocyte infiltration, histological damage and renal function impairment in multiple kidney diseases. MIF was considered to be the early prediction of tissue rejection in experimental and clinical transplantation. MIF is increased in many kidney diseases as: acute kidney injury, lipid-induced glomerular injury, rat crescentic glomerulonephritis, anti-GBM diseases, etc. MIF in plasma and urine is significantly elevated in patients with acute kidney injury (AKI) and elevated MIF in serum is associated with renal function and injury, it represents as a biomarker for renal replacement therapy after AKI. This review provides a brief concept of MIF signaling pathway and functional role of MIF in different kidney disease especially AKI.
Keywords: Macrophage Migration Inhibitory Factor; Acute Kidney Injury; MIF Signaling; Renal Inflammation. EXPRESSION PATTERN OF MIF
Expression of MIF
Macrophage migration inhibitory factor (MIF) was first discovered by Barry Bloom and Boyce Bennett in 1970. They observed a protein from the supernatant of antigen-sensitized lymphocytes could inhibit the migration of macrophages and peritoneal cells [1]. Human MIF cDNA clone and recombinant MIF became available and thus facilitated the analysis of the role of this lymphokine in cellmediated immunity, immunoregulation, and inflammation over decades [2].
MIF expression has been started at the beginning of life. MIF expression has been detected in multiple tissues and different cell types during organogenesis. MIF mRNA expression was detected in somites, precartilage primordia in ribs and vertebrae, branchial arches, limb buds, neural tissues, all muscle cell types and during organogenesis, lung, liver, kidney, testis, spleen, skin, adrenal gland, intestine, adrenal gland and pancreas [3-6]. All tissues express MIF at baseline levels, and it is significantly upregulated under stimulations such as sepsis, stress, or diseased condition. Onset of MIF expression coincides with the specification of tissues, it was observed during myogenesis in all muscle cell types, including cardiac, smooth, and skeletal muscle, during embryonic development [6].
Secretory MIF protein can be detected constitutively in serum and plasma. Historically, MIF was first thought to be produced by activated T-lymphocytes thus considered as a lymphokine, but immunohistochemical analysis of various tissues indicates MIF was also been shown to be secreted from the anterior pituitary gland, monocytes/macrophages, and T and B lymphocytes, NK-cells, basophiles/mast cells and eosinophils activated by various proinflammatory stimuli [7]. MIF was found to be expressed constitutively in anterior pituitary gland, the adrenal cortex, the Leydig-cells of the testis, the epithelial cells of the epididymis and pancreatic β- cells. Other MIF synthesizing cells are vascular smooth muscle, cardiomyocytes and skeletal muscle cells [8], gastric parietal cells [9], keratinocytes and fibroblasts [10], hepatocytes and peripheral and central neurons [4].
Renal MIF is constitutively produced under normal circumstances but significantly upregulated in the kidneyinfiltrating T cells, macrophages and various non-immune cells including tubular and glomerular epithelial cells, mesangial cells, endothelial cells, fibroblasts and vascular smooth muscle cells under diseased conditions. Renal MIF is released and exerts its biological activities in many pathological conditions such as septic shock, renal inflammation, immune injury and diabetes.
Regulation of MIF secretion
MIF is normally released at a low rate and in large amounts after stimulation from leukocytes, immune cells and released by injured cells or dead cells. MIF exists in cytoplasm and release directly after stimulation, which is independent to the endoplasmic reticulum and the Golgi, so, no synthesis is necessary before its release [11,12]. Furthermore, MIF expression may increase after conditions of stimulation as stress, sepsis and hypoxia [13]. Thus, in the MIF secretion curve there are two peaks, the first one is formed by MIF releasing from the cytoplasm stores which is the fast and high peak, and then follows the second peak consequent to new MIF synthesis which is the slow and flat peak. MIF SIGNALING PATHWAY
The receptors of MIF
CD74 is identified as the first and main receptor of MIF [14] and it is an invariant MHC class II cell membrane with highaffinity receptor for bacterial proteins and d-dopachrome tautomerase (d-DT/MIF) [15]. The signals of MIF-CD74 transducing to downstream depends on another receptor CD44, which is a co-receptor of MIF and can interacts with downstream signals as PI3K-Akt, NFκB signaling, etc. CD74 participates in several key processes of the immune system, such as antigen presentation, B-cell differentiation, and inflammatory signaling [16]. MIF and CD74 complex has been shown to regulate peripheral B cell survival. The activation of CD74 also leads to the recruitment of T cells and monocytes, dendritic cell (DC) motility, macrophage inflammation, and thyme selection. CD74 expression was suggested to be as a prognostic factor in many cancers and was suggested to be a predictor of tumor progression [17]. CD74 is a new candidate for immunotherapy of neoplasms, which can be exploited using either a naive anti-CD74 antibody as well as with conjugates including isotopes, drugs, or toxins [18]. CD74 also participates in many human diseases such as inflammatory disease, liver fibrosis, type I diabetes, systemic lupus erythematosus, and Alzheimer disease [19, 20].
Importantly, MIF interacts with extracellular domain of CD74 requires the phosphorylation and the recruitment of CD44 which is a genetically polymorphic molecule with an important role in cell-extracellular matrix interaction to form a MIF/CD74/CD44 complex. It was reported that CD44 play a role in MIF-induced ERK phosphorylation [21]. Study revealed that CD44 deletion partly blocked the inflammatory responses and reduced renal inflammation and injury [22].
Other MIF receptors have been described are the CXC chemokine receptors CXCR2 and CXCR4 which have a relevant role mainly in inflammatory diseases [23]. MIF promotes macrophages and T cells recruit to the inflamed area through binding to CXCR2 and CXCR4. It is also reported that CXCR2 binds to CD74 to form a CD74/CD44/CXCR2 complex which transduce signals downstream.
MIF signaling pathway
MIF has extracellular and intracellular signaling pathways. In extracellular pathway, MIF binds to the extracellular ligands of CD74 which interacts with CD44 to form a complex, than transduce signals to downstream signaling as MAP kinase to stimulate cell proliferation. In addition, MIF also can indirectly activate NF-κB signaling through a signaling cascade including Src kinase, Akt, Syk, which consequently promotes B lymphocytes proliferation and survival. All this signal activation requires CD74 and CD44 receptors participating and this receptors complex is also required in MIF-mediated anti-apoptotic effects. Moreover, MIF binding to its receptor may also due to the activation of ERK1/2-MAPK, JAB1-CSN5, or PI3K-Akt pathways, the inhibition of p53, and the stimulation of antigenic factors including IL-8 and VEGF. In addition, overexpression of MIF also suppresses anti-tumor activity of the host immune system.
For extracellular pathway: MIF is abundantly existed in the cytoplasm and interacts with other signaling pathways. For example, MIF binds to a coactivator of AP-1 transcription Jab-1 (Jun activation domain-binding protein) [24], inhibits Jab1-mediated JNK activation and then enhances c-Jun phosphorylation. MIF also antagonizes JAB dependent cell cycle regulation, which is shown in Figure 1 [24].
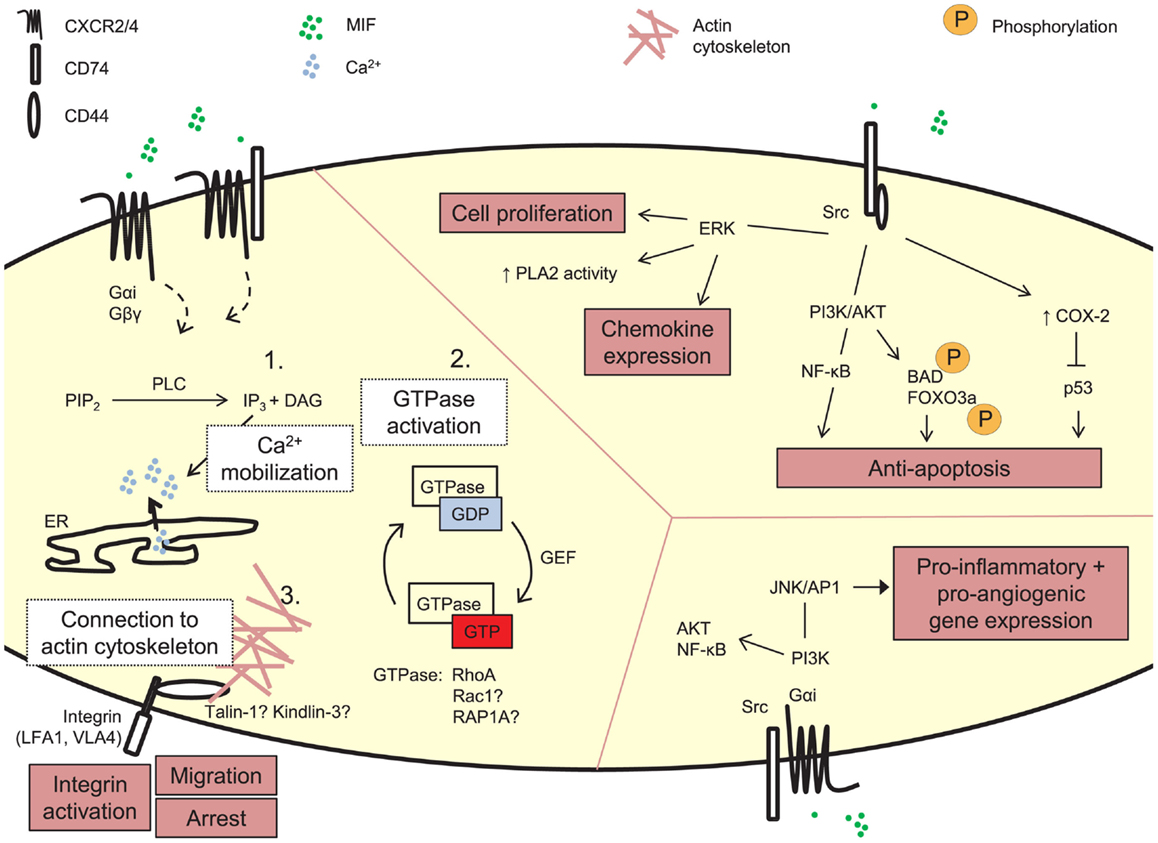
Figure 1: Signaling of MIF. [Sabine Tillmann, Jürgen Bernhagen, Front Immunol., 2013]. MIF IN NON-KIDNEY DISEASES
Macrophage migration inhibitory factor (MIF) has emerged as a promising therapeutic target in human diseases including immune disorder, cancer, cardiologic diseases, diabetes, and inflammatory diseases.
Immune diseases
Increasing evidence shows that MIF is involved in the regulation of T-cell and B-cell developments, dendritic cell (DC) motility, macrophage inflammation, and thymic selection [19]. MIF/CD74 plays an important role in many immune diseases, such as osteoarthritis and rheumatoid arthritis [25], multiple sclerosis [26], Ankylosing spondylitis [27], ANCAassociated vasculitis [28], systemic lupus erythematosus [29]. MIF-deficient MRL/lpr mice have significantly longer survival time and fewer renal and skin injury, where MCP-1 and renal macrophage infiltration were significantly reduced [30]. MIF deletion can protect against disease development of the collagen- and the adjuvant-induced arthritis models [31]. MIF recruits neutrophils via increasing ANCA antigen translocation. The recruited neutrophils can be induced by ANCA further, resulting in respiratory burst and degranulation [28]. MIF is an important element of the inflammatory cascade in rheumatoid arthritis development. Several studies have been demonstrated that the levels of MIF were increased in synovial and serum inRA patients [32,33]. The severity of histological arthritis and cartilage damage, as well as reduced proliferation of synoviocytes was ameliorated in mice lacking MIF [34]. It is also found that MIF promotes leukocyte recruitment in the joint under high endotoxin or TNF condition [35].
Cancers
MIF is recently found to play a prominent role in the cancer progression. Experimental and clinical studies reported that high levels of MIF were observed in a number of human cancers such as Non-small cell lung cancer [36], acute myeloid leukemia [37], breast cancer [38], head and neck squamous cell carcinoma [39], colorectal cancer [40], bladder cancer [41], etc. MIF stimulates the releasing of angiogenic factors that lead to tumor growth and aggressiveness. MIF also triggers the production of cytokines and chemokines in the tumor microenvironments, which suppresses immune surveillance and immune response against tumors, angiogenesis, and carcinogenesis thereby leading to pathological condition e.g. chronic inflammation and immunomodulation [42]. Wu S indicated that MIF promoter polymorphisms (-794CATT) were correlated with the early-stage of cervical cancer [43]. AbdulAziz found that MIF was highly expressed in the primary AML [37]. In addition, MIF can increase the myeloid suppressor cells recruitment and is correlated with bladder cancer via CXCL2/MIF-CXCR2 signaling [41]. Bozzi F revealed that MIF/ CD74 axis can be taken as a new therapeutic target in colon cancers [44].
Cardiologic diseases
MIF is markedly upregulated in vulnerable atheromatous plaques suggests that MIF may be important in the destabilization of human atherosclerotic plaques [45,46]. Upregulation of myocardial MIF was observed and may contribute to macrophage accumulation in the infarcted area and it may play a pro-inflammatory role in the myocyte damage in AMI [47]. Karin A.L. Mueller described that the level of MIF expression is linked to the degree of myocardial fibrosis with progressive chronic HF in patients. MIF predicted allcause mortality and the combined study endpoint [48]. In mice myocyte infarction models, the amelioration of cardiac remodeling and incidence of post-MI cardiac rupture (27% vs. 53%) was much lower in MIF KO mice than MIF WT mice [49].
Diabetes
Recently, the term “meta-inflammation” was used to describe the low-grade systemic inflammation status in diabetes and obesity [50]. Increasing evidence suggests that MIF is involved in meta-inflammatory processes. Many studies described genetic polymorphisms of MIF were associated with increased risk of GDM and insulin resistance in diabetic patients, such as genetic polymorphism of rs755622, rs1007888 in MIF, MIF173GC polymorphism and MIF gene promoter polymorphisms [51-54]. In an animal model of human type 1 diabetes mellitus, MIF was revealed that it plays a critical role in the immunemediated beta-cell destruction [55]. Other study also indicated MIF influences the molecules expression of Mφ and DC activation in T1DM, the expression of MHC-II, costimulatory molecules CD86, CD80, and CD40, TLR-2, and TLR-4 were lower observed in MIF KO mice than MIF WT mice in induced T1DM model [56]. New MIF inhibitors were revealed could reduce inflammation-caused beta cell death [57]. For example, MIF inhibitor ISO-1 was investigated significantly decreased macrophage activation in db/db mice, accompanied with renal function attenuated and the production of inflammatory cytokines reduced [58]. A significant increase of serum level of MIF was found in patients with T1DM which indicated that MIF could be a therapeutic target for diabetes [59].
Inflammatory diseases
MIF is a pleiotropic cytokine which has chemokine-like functions and plays an essential role in both innate and acquired immunity. Dysregulated MIF expression was seen in various inflammatory conditions [60]. The baseline MIF level and inducible MIF expression in the brain reveals the importance role of MIF in inflammatiory response in neuroendocrine system [4]. MIF also plays a role in cellmediated hepatic injury in chronic hepatitis B infection [61]. In addition, H. pylori infection is associated with an increasing of MIF expression in gastric epithelial and inflammatory cells [62]. High expression level of MIF alleles is a genetic marker of morbidity and mortality of pneumococcal meningitis [63]. MIF plays a crucial pathological role sustaining the alveolar inflammatory response in ARDS and that anti-MIF and early glucocorticoid therapy may represent a novel therapeutic approach in inflammatory diseases [64,65]. MIF IN KIDNEY DISEASES
It was reported MIF contributes to leukocyte infiltration, histological damage and renal function impairment in multiple kidney diseases.
MIF is constitutively expressed in normal kidney in macrophages, T and B lymphocytes and various non-immune cells including tubular and glomerular epithelial cells, mesangial cells, endothelial cells, fibroblasts and vascular smooth muscle cells [66,67]. MIF was considered to be the early prediction of tissue rejection in experimental and clinical transplantation [68]. MIF is increased in many kidney diseases as: acute kidney injury, lipid-induced glomerular injury [69], rat crescentic glomerulonephritis [66], anti-GBM diseases [70], ANCN-vasculitis, experimental murine MRL/ lpr lupus nephritis [71], and unilateral ureteral obstruction (UUO) obstructive nephropathy [72], acute renal allograft rejection [73], acute urate nephropathy [74], aristolochic acid nephropathy [75] and IgA nephropathy [76].
In human kidney diseases, dramatic increase in MIF expression is detected in both glomerular and tubular in proliferative glomerulosclerosis, including lupus nephritis, focal segmental glomerulosclerosis (FGS), crescentic glomerulosclerosis, mesangial-capillary proliferative glomerulosclerosis and allograft rejection [67,77]. Furthermore, infiltrating macrophages and T cells also express MIF, Matsumoto, K. described that peripheral blood T cells isolated from patients with IgAN produced more MIF than T cells from healthy controls or patients treated with corticosteroids [78]. Furthermore, urine MIF level in proliferative glomerulonephritis was increased and correlated with the severity of renal injury [79]. Anti-MIF neutralizing antibody treatment or MIF deficiency may protect mice from kidney diseases. Blocking MIF activity with anti-neutralizing antibody can partially reverse mice crescentic glomerulonephritis, suggesting that MIF would enhance the cellular immune response [80]. Blocking MIF using a neutralizing antibody [80] or a MIF inhibitor RPS19 [70] can attenuate renal injury by reducing cytokine production, leukocyte infiltrates and 24-hour proteinuria in anti-GBM glomerulonephritis. In another research of a mouse model of IgAN, anti-MIF treatment can ameliorate kidney injury and reduce renal TGF-β1 expression [81]. ACUTE KIDNEY INJURY
Pathophysiology of Acute Kidney Injury
Acute kidney injury (AKI) is one of the causes leading to chronic kidney diseases (CKD) and is related with high mortality rates. The main causes of AKI including prolonged renal ischemia, nephrotoxins, glomerular diseases, obstructed ultrafiltration. The characteristic of AKI is rapidly declined in GFR. Inflammation is an important additional phenomenon of AKI exuberate kidney injury. Renal injury usually affects the highly metabolic active nephron segments in the renal outer medulla, which more likely to suffer kidney injury as reversible conditions of hypoxia to intrinsic renal failure. The essence of the recovery is the injured tubular epithelial cells can restore to normal function and promote regeneration. Recent researchers suggest that AKI has a potential tendency to CKD. Thus, early diagnosis of AKI is essential for treating patients with AKI, and potential biomarkers of AKI may be a promising therapeutic target in the future.
Inflammation and acute kidney injury
Inflammation is the main characteristic of AKI. Inflammation is a significant component of renal I/R injury, playing a considerable role in its pathophysiology. Endothelial injury, generation of inflammatory mediators, leukocyte infiltration largely contributes to the pathogenesis of AKI.
Injury of the kidney contributes to inflammatory response, results in endothelial activation and injury, enhances leukocyte entrapment, endothelial cell-leukocyte adhesion and an accommodation in microvascular blood flow as shown in Figure 2 [82,83].
The outer medulla is impacted in a greater extent than the cortex during leukocyte-endothelial, which is indicated by the marked vascular overcrowding seen in the outer medulla. Leukocyte subgroups such as neutrophils and T lymphocytes are all contributed to I/R injury [84-86]. Neutrophils from patients with sepsis-induced AKI showed abolished exvivo slow rolling, compared with neutrophils from healthy volunteers and patients with sepsis but no AKI. Blocking neutrophil infiltration protects the kidney against ischemic renal injury, even when the antibody was administered after ischemic happened [87]. CD4/CD8 Knockout mice are protected against I/R injury, with a reduction of neutrophils infiltration and T cells adhesion to the renal tubular epithelial cells, suggesting a pathophysiological role for neutrophils and T lymphocytes in AKI [88]. Macrophages infiltrate the injured kidney within 1 hour of ischemia reperfusion and this activity is mediated by fractalkine (CXCL1), both ischemiaand cisplatin-induced AKI triggers fractalkine expression in peritubular capillary endothelial cells. Using anti-CX3C receptor-1 antibody can effectively attenuate the severity of AKI in mice, macrophages lacking CCR do not infiltrate injured kidneys and the resultant injury is less severe[89]; while transferring activated RAW 264.7 macrophages exacerbates kidney injury [89,90].
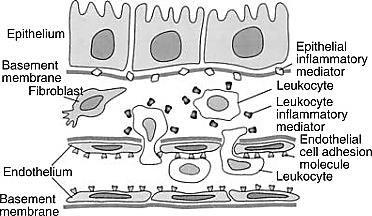
Figure 2: Schematic illustration of the inflammatory mediators produced by tubular epithelial cells and activated leukocytes in renal ischemia/ reperfusion (I/R) injury [84] (Joseph V. Bonventre & Anna Zuk, Kidney Int., 2004).
The outer medulla is impacted in a greater extent than the cortex during leukocyte-endothelial, which is indicated by the marked vascular overcrowding seen in the outer medulla. Leukocyte subgroups such as neutrophils and T lymphocytes are all contributed to I/R injury [84-86]. Neutrophils from patients with sepsis-induced AKI showed abolished exvivo slow rolling, compared with neutrophils from healthy volunteers and patients with sepsis but no AKI. Blocking neutrophil infiltration protects the kidney against ischemic renal injury, even when the antibody was administered after ischemic happened [87]. CD4/CD8 Knockout mice are protected against I/R injury, with a reduction of neutrophils infiltration and T cells adhesion to the renal tubular epithelial cells, suggesting a pathophysiological role for neutrophils and T lymphocytes in AKI [88]. Macrophages infiltrate the injured kidney within 1 hour of ischemia reperfusion and this activity is mediated by fractalkine (CXCL1), both ischemiaand cisplatin-induced AKI triggers fractalkine expression in peritubular capillary endothelial cells. Using anti-CX3C receptor-1 antibody can effectively attenuate the severity of AKI in mice, macrophages lacking CCR do not infiltrate injured kidneys and the resultant injury is less severe[89]; while transferring activated RAW 264.7 macrophages exacerbates kidney injury [89,90].
In addition to the accumulation of leukocytes and endothelial cells injury to the inflammatory response in AKI, the injured tubular epithelial cells and activated leukocytes also generate mediators that exacerbate inflammation including TNF-α, IL1, IL-6, IL-8, TGF-β, MCP-1, ENA-78, RANTES, and fractalkines [91]; while leukocytes may produce IL-1, IL8, MCP-1, reactive oxygen species and eicosanoids. Both experimental and clinical data have been shown that AKI exerts its regulatory effects on innate immunity via modulating the cytokine homeostasis [92]. In mice model of AKI, the surgery leads to a profound release of proinflammatory cytokines (IL-6 or TNF-α), and remain increased for several days. While sham surgery does not lead to such a prolonged cytokine release [93]. Study indicated TLR4 may also very important at the beginning of transplant. Less MCP-1 and TNFα were detected in the donor kidneys in TLR4 deletion mice but with more heme-oxygenase 1 (HO-1) expression [94]. The decline in renal function during AKI is likely to play a major role in cytokine clearance in a rat model of rhabdomyolysis-induced AKI [95]. This decline in turn led to a sustained increase in plasma cytokine concentrations. Another mechanism cytokine levels are increased in AKI may be augmented production of inflammatory mediators by renal tubular cells in response to injury or cytokines [96,97]. Specific cytokine intervention may offer a new therapeutic hope.
Although significant progress has been made in defining the major components of MIF-mediated AKI, the complex cross talk between endothelial cells, inflammatory cells, and the injured epithelium with each generating and responding to cytokines and chemokines is not well understood. MIF ASSOCIATED RENAL INFLAMMATION IN AKI
Elevated urinary MIF has previously been observed in AKI during kidney infection in patients, and accompanied with the severity of renal injury in acute pyelonephritis, Brown FG indicated that urine MIF concentration is correlated with the degree of renal dysfunction, histologic damage, and leukocytic infiltration in human glomerulonephritis and has also been suggested as a potential biomarker for acute kidney damage [98,99]. Similar findings have been exhibited in kidney transplantation, urinary MIF was increased on day 1 posttransplantation and changed parallel with the serum creatinine, urine MIF increased even before biopsy proven acute renal rejection [79]. “Severe AKI” had higher levels of plasma IL-10, MIF and IL-6 compared to “no AKI” and “mild AKI” in septic patients admitted to the ICU [100]. In animal models and in vitro studies, MIF induced leukocytes accumulation as well as tissue infiltration of leukocytes thus induces multi-organ damage affecting both lungs and kidneys, treatment with antiMIF antibody attenuated pulmonary pathology in mice with LPS-induced acute lung injury or anti-GBM glomerulonephritis via reducing the upregulation of IL-1β, ICAM-1 and VCAM-1 [101-103]. Recently study reported that plasma MIF level in was elevated in AKI patients after orthotropic liver transplantation patients and was more valuable in identifying the prognosis of AKI and predicting the requirements of renal replacement therapy after operation [104]. MIF and its receptor CD74 may be useful targets to reduce neutrophilic inflammation in acute lung injury [101]. In heart, the upregulation of MIF levels contributes to AMPK activation thus protects mice from heart infarction [105]. The mechanism of MIF in the progression of AKI is needed to be further elucidated.
Acknowledgements
The Study is supported by NSFC Funds in China NSFC 81900673, JCYJ20180307150634856. REFERENCES
- Bloom, B.R. and B. Bennett. (1970). Relation of the migration inhibitory factor (MIF) to delayed-type hypersensitivity reactions. Ann N Y Acad Sci. 169(1): 258-265.
- Weiser WY, Temple PA, Witek-Giannotti JS, Remold HG et al. (1989).Molecular cloning of a cDNA encoding a human macrophage migration inhibitory factor. Proc Natl Acad Sci U S A. 86(19): 7522-7526.
- Suzuki HH, Kanagawa and J. Nishihira. (1996). Evidence for the presence of macrophage migration inhibitory factor in murine reproductive organs and early embryos. Immunol Lett. 51(3): 141-147.
- Bacher M, Meinhardt A, Lan HY, Dhabhar FS, et al. (1998). MIF expression in the rat brain: implications for neuronal function. Mol Med. 4(4): 217-230.
- Suzuki T, Ogata A, Tashiro K, Nagashima K et al. (1999).Augmented expression of macrophage migration inhibitory factor (MIF) in the telencephalon of the developing rat brain. Brain Res. 816(2): 457- 462.
- Kobayashi S, Satomura K, Levsky JM, Sreenath T, et al. (1999).Expression pattern of macrophage migration inhibitory factor during embryogenesis. Mech Dev. 84(1-2): 153-156.
- Bacher M, Meinhardt A, Lan HY, Mu W, et al. (1997). Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol. 150(1): 235-246.
- Benigni F, Atsumi T, Calandra T, Metz C, et al. (2000). The proinflammatory mediator macrophage migration inhibitory factor induces glucose catabolism in muscle. J Clin Invest. 106(10): 1291-1300.
- Kudo T. (1998). [Identification of macrophage migration inhibitory factor (MIF) in rat gastrointestinal tract and its role in rat stomach]. Hokkaido Igaku Zasshi. 73(4): 317-325.
- Abe R, Tadamichi Shimizu, Akira Ohkawara and Jun Nishihira. (2000) Enhancement of macrophage migration inhibitory factor (MIF) expression in injured epidermis and cultured fibroblasts. Biochim Biophys Acta. 1500(1): 1-9.
- Merk M, Baugh J, Zierow S, Leng L, et al. (2009). The Golgi-associated protein p115 mediates the secretion of macrophage migration inhibitory factor. J Immunol. 182(11): 6896-6906.
- Merk M, Mitchell RA, Endres S, Bucala R, et al. (2012). D-dopachrome tautomerase (D-DT or MIF-2): doubling the MIF cytokine family. Cytokine. 59(1): 10-17.
- Simons D, Grieb G, Hristov M, Pallua N, et al. (2011). Hypoxia-induced endothelial secretion of macrophage migration inhibitory factor and role in endothelial progenitor cell recruitment. J Cell Mol Med. 15(3): 668-678.
- Leng L, Metz CN, Fang Y, Xu J, et al. (2003). MIF signal transduction initiated by binding to CD74. J Exp Med. 197(11): 1467-1476.
- Valino-Rivas L, Baeza-Bermejillo C, Gonzalez-Lafuente L, Sanz AB, et al. (2015). CD74 in Kidney Disease. Front Immunol. 6: 483.
- Borghese F and Clanchy FI. (2011). CD74: an emerging opportunity as a therapeutic target in cancer and autoimmune disease. Expert Opin Ther Targets. 15(3): 237-251.
- Shachar I and M Haran. (2011). The secret second life of an innocent chaperone: the story of CD74 and B cell/chronic lymphocytic leukemia cell survival. Leuk Lymphoma. 52(8): 1446-1454.
- Stein R, Mattes MJ, Cardillo TM, Hansen HJ, et al. (2007). CD74: a new candidate target for the immunotherapy of B-cell neoplasms. Clin Cancer Res. 13(18 Pt 2): 5556s-5563s.
- Su H, Na N, Zhang X, Zhao Y, et al. (2017). The biological function and significance of CD74 in immune diseases. Inflamm Res. 66(3): 209-216.
- Bucala R and I Shachar. (2014). The integral role of CD74 in antigen presentation, MIF signal transduction, and B cell survival and homeostasis. Mini Rev Med Chem. 14(14): 1132-1138.
- Shi X, Leng L, Wang T, Wang W, et al. ( 2006). CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity. 25(4): 595-606.
- Rampanelli E, Mark C, Dessing, Nike Claessen, Gwendoline JD Teske, et al. (2013). CD44-deficiency attenuates the immunologic responses to LPS and delays the onset of endotoxic shock-induced renal inflammation and dysfunction. PLoS One. 8(12): e84479.
- Bernhagen J, Krohn R, Lue H, Gregory JL, et al. (2007). MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 13(5): 587-596.
- Tillmann S, J Bernhagen, and H Noels. (2013). Arrest Functions of the MIF Ligand/Receptor Axes in Atherogenesis. Front Immunol. 4: 115.
- Kim K W and H R Kim. (2016). Macrophage migration inhibitory factor: a potential therapeutic target for rheumatoid arthritis. Korean J Intern Med. 31(4): 634-642.
- Di Marco J, De Broglie C, Manceau, Weglinski L, et al. (2016). [Anticholinergics do not affect the sexual function in women diagnosed with multiple sclerosis]. Prog Urol. 26(4): 226-229.
- 27. Gurel C, Ahmet İnanır, Ayşe Feyda Nursa, Aydin Rustemoglu, et al.( 2016). Evaluation of MIF -173 G/C Polymorphism in Turkish Patients with Ankylosing Spondylitis. Balkan Med J. 33(6): 614-619.
- Hao J, Lv TG, Wang C, Xu LP, et al. (2016). Macrophage migration inhibitory factor contributes to anti-neutrophil cytoplasmic antibody-induced neutrophils activation. Hum Immunol. 77(12): 1209-1214.
- Connelly K L, Kandane-Rathnayake R, Hoi A, Nikpour M, et al. (2016). Association of MIF, but not type I interferon-induced chemokines, with increased disease activity in Asian patients with systemic lupus erythematosus. Sci Rep. 6: 29909.
- Hoi AY, Hickey MJ, Hall P, Yamana J, et al. (2006). Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. J Immunol. 177(8): 5687-5696.
- Greven D, Leng L and Bucala R. (2010). Autoimmune diseases: MIF as a therapeutic target. Expert Opin Ther Targets. 14(3): 253-264.
- Radstake TR, Sweep FC, Welsing P, Franke B, et al. (2005). Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum. 52(10): 3020-3029.
- Leech M, Metz C, Hall P, Hutchinson P, Gianis K, et al. (1999). Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 42(8): 1601-1608.
- Leech M, Lacey D, Xue JR, Santos L, et al. (2003). Regulation of p53 by macrophage migration inhibitory factor in inflammatory arthritis. Arthritis Rheum. 48(7): 1881-1889.
- Gregory JL, Leech MT, David JR, Yang YH, et al. (2004). Reduced leukocyte-endothelial cell interactions in the inflamed microcirculation of macrophage migration inhibitory factor-deficient mice. Arthritis Rheum. 50(9): 3023-3034.
- Eide HA, Halvorsen AR, Sandhu V, Fane A, et al. (2016). Non-small cell lung cancer is characterised by a distinct inflammatory signature in serum compared with chronic obstructive pulmonary disease. Clin Transl Immunology. 5(11): e109.
- Abdul-Aziz AM, Shafat MS, Mehta TK, Di Palma F, et al. (2017). MIF-Induced Stromal PKCbeta/IL8 Is Essential in Human Acute Myeloid Leukemia. Cancer Res. 77(2): 303-311.
- Lin S, Wang M, Liu X, Zhu W, et al. (2016). Association of genetic polymorphisms in MIF with breast cancer risk in Chinese women. Clin Exp Med. 17(3): 395-401.
- Wang SS, Cen X, Liang XH and Tang YL. (2016). Macrophage migration inhibitory factor: a potential driver and biomarker for head and neck squamous cell carcinoma. Oncotarget. 8(6): 10650-10661.
- Park GB, Chung YH, Gong JH, Jin DH, et al. (2016). GSK-3beta-mediated fatty acid synthesis enhances epithelial to mesenchymal transition of TLR4-activated colorectal cancer cells through regulation of TAp63. Int J Oncol. 49(5): 2163-2172.
- Zhang H, Ye YL, Li MX, Ye SB, et al. (2016). CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene. 36(15): 2095-2104.
- Nobre CC, de Araujo JM, Fernandes TA, Cobucci RN, et al. (2016). Macrophage Migration Inhibitory Factor (MIF): Biological Activities and Relation with Cancer. Pathol Oncol Res. 23(2): 235-244.
- Wu S, Sun J, Lian J, Shang H, et al. (2017). Macrophage migration inhibitory factor promoter polymorphisms (-794CATT5-7) as potential biomarker for early-stage cervical cancer. J Obstet Gynaecol Res. 43(3): 571-579.
- Bozzi F, Mogavero A, Varinelli L, Belfiore A, et al. (2017). MIF/CD74 axis is a target for novel therapies in colon carcinomatosis. J Exp Clin Cancer Res. 36(1): 16.
- Kong YZ, Huang XR, Ouyang X, Tan JJ, et al. (2005). Evidence for vascular macrophage migration inhibitory factor in destabilization of human atherosclerotic plaques. Cardiovasc Res. 65(1): 272-282.
- Kong YZ, Yu X, Tang JJ, Ouyang X, et al. (2005). Macrophage migration inhibitory factor induces MMP-9 expression: implications for destabilization of human atherosclerotic plaques. Atherosclerosis. 178(1): 207-215.
- Yu CM, Lai KW, Chen YX, Huang XR, et al. (2003). Expression of macrophage migration inhibitory factor in acute ischemic myocardial injury. J Histochem Cytochem. 51(5): 625-631.
- Mueller KA, Schwille J, Vollmer S, Ehinger E, et al. (2016). Prognostic impact of macrophage migration inhibitory factor in patients with non-ischemic heart failure undergoing endomyocardial biopsy. Int J Cardiol. 203(1): 656-659.
- White DA, Su Y, Kanellakis P, Kiriazis H, et al. (2014). Differential roles of cardiac and leukocyte derived macrophage migration inhibitory factor in inflammatory responses and cardiac remodelling post myocardial infarction. J Mol Cell Cardiol. 69(1): 32-42.
- Leon-Pedroza JI, Gonzalez-Tapia LA, del Olmo-Gil E, Castellanos-Rodriguez D, et al. (2015). [Low-grade systemic inflammation and the development of metabolic diseases: from the molecular evidence to the clinical practice]. Cir Cir. 83(6): 543-551.
- Li C, Qiao B, Qi W, Zhan Y, et al. (2016). Association of Macrophage Migration Inhibitory Factor Polymorphisms with Gestational Diabetes Mellitus in Han Chinese Women. Gynecol Obstet Invest. 81(1): 84-89.
- Ines Matia-Garcia, Lorenzo Salgado-Goytia, Jose F, Munoz-Valle I, et al. (2015). Macrophage migration inhibitory factor promoter polymorphisms (-794 CATT 5-8 and -173 G>C): relationship with mRNA expression and soluble MIF levels in young obese subjects. Dis Markers. 2015: 461208.
- Zhan Y, Li C, Chen J, Yu S, et al. (2015). Association between macrophage migration inhibitory factor rs1007888 and GDM. Genet Mol Res. 14(1): 797-804.
- Coban N, Onat A, Yildirim O, Can G, et al. (2015). Oxidative stress-mediated (sex-specific) loss of protection against type-2 diabetes by macrophage migration inhibitory factor (MIF)-173G/C polymorphism. Clin Chim Acta. 438: 1-6.
- Cvetkovic I, Al-Abed Y, Miljkovic D, Maksimovic-Ivanic, et al. (2005). Critical role of macrophage migration inhibitory factor activity in experimental autoimmune diabetes. Endocrinology.146(7): 2942-2951.
- Morsi HK, Ismail MM, Gaber HA, and Elbasmy AA. (2016). Macrophage Migration Inhibitory Factor and Malondialdehyde as Potential Predictors of Vascular Risk Complications in Type 2 Diabetes Mellitus: Cross-Sectional Case Control Study in Saudi Arabia. Mediators Inflamm. 2016: 5797930.
- Vujicic M, Nikolic I, Krajnovic, Cheng KF, et al. (2014). Novel inhibitors of macrophage migration inhibitory factor prevent cytokine-induced beta cell death. Eur J Pharmacol. 740: 683-689.
- Wang Z, Wei M, Wang M, Chen L, et al. (2014). Inhibition of macrophage migration inhibitory factor reduces diabetic nephropathy in type II diabetes mice. Inflammation. 37(6): 2020-2029.
- Ismail NA, Abd El Baky AN, Ragab S, Hamed M, et al. (2016). Monocyte chemoattractant protein 1 and macrophage migration inhibitory factor in children with type 1 diabetes. J Pediatr Endocrinol Metab. 29(6): 641-645.
- Asare Y, M Schmitt, and J Bernhagen. (2013). The vascular biology of macrophage migration inhibitory factor (MIF). Expression and effects in inflammation, atherogenesis and angiogenesis. Thromb Haemost. 109(3): 391-398.
- Zhang HY, Nanji AA, Luk JM, Huang XR, et al. (2005). Macrophage migration inhibitory factor expression correlates with inflammatory changes in human chronic hepatitis B infection. Liver Int. 25(3): 571-579.
- Xia HH, Lam SK, Huang XR, Wong WM, et al. 2004. Helicobacter pylori infection is associated with increased expression of macrophage migratory inhibitory factor--by epithelial cells, T cells, and macrophages--in gastric mucosa. J Infect Dis. 190(2): 293-302.
- Savva A, Brouwer MC, Roger T, Valls Seron M, et al. (2016). Functional polymorphisms of macrophage migration inhibitory factor as predictors of morbidity and mortality of pneumococcal meningitis. Proc Natl Acad Sci U S A. 113(13): 3597-3602.
- Donnelly SC, Haslett C, Reid PT, Grant IS, et al. (1997). Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med. 3(3): 320-323.
- Guo Y and C Xie. (2002). [The pathogenic role of macrophage migration inhibitory factor in acute respiratory distress syndrome]. Zhonghua Jie He He Hu Xi Za Zhi. 25(6): 337-340.
- Lan HY, Mu W, Yang N, Meinhardt A, et al. (1996). De Novo renal expression of macrophage migration inhibitory factor during the development of rat crescentic glomerulonephritis. Am J Pathol. 149(4): 1119-1127.
- Lan HY, Yang N, Nikolic-Paterson DJ, Yu XQ, et al. (2000). Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int. 57(2): 499-509.
- Sakagami K. (1976). Early prediction of acute rejection after inbred rat kidney transplantation using macrophage migration inhibition test. Acta Med Okayama. 30(3): 181-195.
- Miyazaki K, Isbel NM, Lan HY, Hattori M, et al. (1997). Up-regulation of macrophage colony-stimulating factor (M-CSF) and migration inhibitory factor (MIF) expression and monocyte recruitment during lipid-induced glomerular injury in the exogenous hypercholesterolaemic (ExHC) rat. Clin Exp Immunol. 108(2): 318-323.
- Lv J, Huang XR, Klug J, Frolich S, et al. (2013). Ribosomal protein S19 is a novel therapeutic agent in inflammatory kidney disease. Clin Sci (Lond). 124(10): 627-637.
- Leng L, Chen L, Fan J, Greven D, et al. (2011). A small-molecule macrophage migration inhibitory factor antagonist protects against glomerulonephritis in lupus-prone NZB/NZW F1 and MRL/lpr mice. J Immunol. 186(1): 527-538.
- Rice EK, Nikolic-Paterson DJ, David JR, Bucula R, et al. (2004). Macrophage accumulation and renal fibrosis are independent of macrophage migration inhibitory factor in mouse obstructive nephropathy. Nephrology (Carlton). 9(5): 278-287.
- Brown FG, Nikolic-Paterson DJ, Metz C, Bucula R, et al. (1999). Up-regulation of macrophage migration inhibitory factor in acute renal allograft rejection in the rat. Clin Exp Immunol. 118(2): 329-336.
- Kim YG, Huang XR, Suga S, Mazzali M, et al. (2000). Involvement of macrophage migration inhibitory factor (MIF) in experimental uric acid nephropathy. Mol Med. 6(10): 837-848.
- Dai XY, Xiao R Huang, Li Zhou, Lin Zhang, et al. (2016). Targeting c-fms kinase attenuates chronic aristolochic acid nephropathy in mice. Oncotarget. 7(10): 10841-10856.
- Leung JC, Tang SC, Chan LY, Tsang AW, et al. (2003). Polymeric IgA increases the synthesis of macrophage migration inhibitory factor by human mesangial cells in IgA nephropathy. Nephrol Dial Transplant. 18(1): 36-45.
- Lan HY, Yang N, Brown FG, Isbel NM, et al. (1998). Macrophage migration inhibitory factor expression in human renal allograft rejection. Transplantation. 66(11): 1465-1471.
- Matsumoto K and K. Kanmatsuse. (2001). Increased production of macrophage migration inhibitory factor by T cells in patients with IgA nephropathy. Am J Nephrol. 21(6): 455-464.
- Brown FG, Nikolic-Paterson DJ, Chadban SJ, Dowling J, et al. (2001). Urine macrophage migration inhibitory factor concentrations as a diagnostic tool in human renal allograft rejection. Transplantation. 71(12): 1777-1783.
- Yang N, Nikolic-Paterson DJ, Ng YY, Mu W, et al. (1998). Reversal of established rat crescentic glomerulonephritis by blockade of macrophage migration inhibitory factor (MIF): potential role of MIF in regulating glucocorticoid production. Mol Med. 4(6): 413-424.
- Leung JC, Chan LY, Tsang AW, Liu EW, et al. (2004). Anti-macrophage migration inhibitory factor reduces transforming growth factor-beta 1 expression in experimental IgA nephropathy. Nephrol Dial Transplant. 19(8): 1976-1985.
- Linas SL, Shanley PF, Whittenburg D, Berger E, et al. (1988). Neutrophils accentuate ischemia-reperfusion injury in isolated perfused rat kidneys. Am J Physiol. 255(4 Pt 2): F728-735.
- Willinger CC, Schramek H, Pfaller K and Pfaller W. (1992). Tissue distribution of neutrophils in postischemic acute renal failure. Virchows Arch B Cell Pathol Incl Mol Pathol. 62(4): 237-243.
- Bonventre JV and A Zuk. (2004). Ischemic acute renal failure: an inflammatory disease? Kidney Int. 66(2): 480-485.
- Hellberg PO and TO Kallskog. (1989). Neutrophil-mediated post-ischemic tubular leakage in the rat kidney. Kidney Int. 36(4): 555-561.
- Klausner JM, Paterson IS, Goldman G, Kobzik L, et al. (1989). Postischemic renal injury is mediated by neutrophils and leukotrienes. Am J Physiol. 256(5 Pt 2): F794-F802.
- Kelly KJ, Williams WW Jr, Colvin RB and Bonventre JV. (1994). Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proc Natl Acad Sci U S A. 91(2): 812-816.
- Rabb H, Daniels F, O'Donnell M, Haq M et al. (2000). Pathophysiological role of T lymphocytes in renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol. 279(3): F525-F531.
- Li L, Huang L, Sung JS, Vergis AL, et al. (2008). The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 74(12): 1526-1537.
- Oh D J, Dursun B, He Z, Lu L, et al. (2008). Fractalkine receptor (CX3CR1) inhibition is protective against ischemic acute renal failure in mice. Am J Physiol Renal Physiol. 294(1): F264-F271.
- Safirstein R, Megyesi J, Saggi SJ, Price PM, et al. (1991). Expression of cytokine-like genes JE and KC is increased during renal ischemia. Am J Physiol. 261(6 Pt 2): F1095-F1101.
- Singbartl K and M Joannidis. (2015). Short-term Effects of Acute Kidney Injury. Crit Care Clin. 31(4): 751-762.
- Maatman RG, van de Westerlo EM, van Kuppevelt TH and Veerkamp JH. (1992). Molecular identification of the liver- and the heart-type fatty acid-binding proteins in human and rat kidney. Use of the reverse transcriptase polymerase chain reaction. Biochem J. 288( Pt 1): 285-290.
- Kruger B, Krick S, Dhillon N, Lerner SM, et al. (2009). Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci U S A. 106(9): 3390-3395.
- Yamamoto T, Noiri E, Ono Y, Doi K, et al. (2007). Renal L-type fatty acid--binding protein in acute ischemic injury. J Am Soc Nephrol. 18(11): 2894-2902.
- Yamashita T, Doi K, Hamasaki Y, Ishii T, et al. (2014). Evaluation of urinary tissue inhibitor of metalloproteinase-2 in acute kidney injury: a prospective observational study. Crit Care. 18(6): 716.
- Kjeldsen L, Johnsen AH, Sengelov H and Borregaard N. (1993). Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 268(14): 10425-10432.
- Brown FG, Nikolic-Paterson DJ, Hill PA, Isbel NM, et al. (2002). Urine macrophage migration inhibitory factor reflects the severity of renal injury in human glomerulonephritis. J Am Soc Nephrol 13(Suppl 1): S7-13.
- Hong MY, Chin-Chung Tseng, Sheng-Hsiang Lin, Chiou-Feng Lin, et al. (2012). Urinary macrophage migration inhibitory factor serves as a potential biomarker for acute kidney injury in patients with acute pyelonephritis. Mediators Inflamm 2012: 381358.
- Payen D, Busson M, Loiseau P, Legrand M, et al. (2012). A multicentre study of acute kidney injury in severe sepsis and septic shock: association with inflammatory phenotype and HLA genotype. PLoS One7(6): e35838.
- Takahashi K, Koga K, Zhang Y, OJamaa K, et al. (2009). Macrophage CD74 contributes to MIF-induced pulmonary inflammation. Respir Res. 10: 33.
- Lai KN, Leung JC, Metz CN, Bucala R, et al. (2003). Role for macrophage migration inhibitory factor in acute respiratory distress syndrome. J Pathol. 199(4): 496-508.
- Lan HY, Bacher M, Yang N, Mu W, et al. (1997). The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J Exp Med. 185(8): 1455-1465.
- Stefaniak J, Miller EJ, Baron D, Krenn CG, et al. (2015). Macrophage migration inhibitory factor as a potential predictor for requirement of renal replacement therapy after orthotopic liver transplantation. Liver Transpl. 21(5): 662-669.
- Mount PF, Gleich K, Tam S, Fraser SA, et al. (2012). The outcome of renal ischemia-reperfusion injury is unchanged in AMPK-beta1 deficient mice. PLoS One. 7(1): e29887.