Previous Issues Volume 1, Issue 1 - 2016
The Long Journey of Cancer Modeling: Ubi Sumus? Quo Vadimus?
Ugo Rovigatti1
Department of Experimental and Clinical Medicine, University of Florence, Florence, Italy.
Corresponding Author: Ugo Rovigatti, Department of Experimental and Clinical Medicine, University of Florence, Florence, Italy, Tel: +0039-389 5608777; E-Mail: [email protected]
Received Date: 12 Jan 2016 Accepted Date: 19 Jan 2016 Published Date: 23 Jan 2016
Copyright © 2016 Ugo R
Citation: Ugo R. (2016). The Long Journey of Cancer Modeling: Ubi Sumus? Quo Vadimus? Mathews J Cancer Sci. 1(1): 001.
KEYWORDS
NGS Technologies; Cancer Modeling; Cancer Genetics (CAN-GEN); Cancer Upstream (UP-CAN); Genome-Snipers.
INTRODUCTION
In a recent review article, an analysis began of the current status of research in the so-called Next Generation Sequencing Era (NGSE) [1]. There are several reasons for this efforts and three will be summarized in this Mini-Review: 1. The technological advances leading to ever faster and less expensive sequencing methods for cancer patients [2-4]; 2. The logical conclusion that we are reaching an "end-of-the-road" situation in our understanding of the molecular basis of cancer [5, 6]; and 3. The need to rationalize and enormously growing field and to identify some priorities for future successful interventions [1, 7-9].
1. Technological Advances:
These are witnessed even in the short span of a few months [1]. Although the leading and most utilized Illumina and IonTorrent technologies maintain their prime role on the screen, great progresses are also witnessed for the emerging nanopore technology [10-14]. As previously mentioned, Oxford Nanopore Technologies (ONT) has recently launched the Mini-Ion system for rapid, easy and long-range sequencing [15]. For sequence quality, a number of members of Mini-Ion Assess Program (MAP) started evaluating the instrumentation outputs in May 2014 [1, 14]. ONT still requires several optimizations as previously indicated [1, 16]. One of the major problems, still unresolved, of NP technology is the presence of intrinsically high error-rate. This is generally evaluated in the order of 30%, while different assessment have spanned between 5% and 40% [14, 17]. Although this problem is being addressed and reasons beginning to be understood – probably due to some unspecific binding, oscillation of the nucleic acid at the pore entrance and in binding a-haemolysin protein as well as nano-amperage reading pattern ambiguity [1] –, high error rate is hampering direct utilization of NP technology for genome sequencing. While alternative solutions are being considered with MspA protein, which may be more efficient and specific [18-20], most of today's methods rely on: A. Correcting algorithms and soft-wares and B. Parallel readings with the state of the art technology (Illumina or Ion Torrent).
Bioinformatic tools convert the amperage change in nucleotide sequence (basecalling): standard ONT software allows double readings (2 directional) into FASTA5 format [21], then extracted by programs such as PORETOOLS or PoRe into FASTA [22, 23]. The problem of alignment has been typically addressed by programs such as LAST, BLASR, BWA-MEM and margin-Align. [24-27]. The question of monitoring the readouts and alignments is essential with such a high error rate and tools are becoming available: the minoTour and most recently the Nano-OK , which allows alignment based quality control and estimate of error-rate, as well as the Nanocorr algorithm, which specifically corrects the NP readouts [14, 28, 29]. 2. Parallel readings however still seems to be a pursued strategy, in order to obtain meaningful sequences. This has become standard practice both for error correction and for assembling genomic sequences. Recently, the group of McCombie at CSHL has tested the MinION ONT platform for sequencing and assembling the Saccharomyces Cerevisiae genome with parallel sequencing performed with MiSeq (Illumina) [14]. Only by performing correction with the previously mentioned Nanocorr soft-ware, were they able of obtaining –by comparison with MiSeq shorter sequences- a complete and accurate assembly of yeast genome. A similar analysis was also performed with data (sequences) from E. coli [14, 21]. Generally speaking, the NP technology reads are much longer than with Illumina/Ion Torrent and so are the contigs (678 kb versus 59.9 Kb, i.e. approximately 10X magnification). This is certainly one of its most important qualities (once the error issue will be solved), essential for efficient sequencing of novel genomes [14]. This very short photogram of NP technology at 2016 incipit can just give an idea of the fast-pace of this evolving field. It is however foreseeable that we will have much more efficient and less expensive technologies –already approaching the $1000 human genome goal of G. Church- in the years/months to come [1, 30]. The next and real questions are: how far do have to keep improving sequencing for understanding cancer cell? Are we moving in the right direction, or better: today's cancer genomics has only one possible explanation?
2. End-Of-The-Road" (Eor)
For the second question, how far can we reasonably keep searching before reaching the so-called "end-of-theroad" (EOR), even without crystal balls some reasonable consideration can be made [1]. Searching for the "cancergenome" is reminiscent of what happened in the 50'-60', when molecular biologists were searching "for the gene". Then, great Pioneers such as Jonathan Beckwith, James Shapiro, Saymour Benzer and many others were capitalizing from previous work of Morgan, McClintock, Beadle, Tatum, Lederberg, Watson, Crick, Jacob, Monod and others for finally identifying the entity molecular biologists considered their Saint-Graal: "the gene" [31]. However, it became immediately clear already from the work of S. Benzer that the end-of-the-road was going to be reached soon [32]. Benzer unequivocally demonstrated in his study of the RII region of phage T4 -already at the end of the 50's- that the gene had a defined structure, clearly identifiable by thousands of recombination events [32, 33]. The first gene, the Lac Operon finally isolated and visualized for the first time by the Harvard team of Beckwith and Shapiro [34], was already clearly delineated in the experiments of Benzer over 10 years earlier [32]. Recombination (and later complementation) had delineated an inescapable path toward definition of gene structure. Or to put it differently, the genetic analysis could not proceed any further or to a finer level than what Benzer had done [32-34]. Similarly today, NGS analysis is bringing us to another end-of-road (EOR). Becoming capable of analyzing the entire genome of theoretically any cancer cell will lead us to the full understanding of cancer cells ? Genetically, certainly yes: there is not additional or more sophisticated analysis that we can do. Yet, the answer(s) for cancer understanding may be different from what expected [1, 5]. For some years now, the paradigm "cancer is genetic" has dominated the research field. Unquestionably, the seminal paper by Hanahan and Weinberg on Hallmarks of Cancer (HoC) at the end of last Century (and Millennium) has paved the way for a robust compendium of cancer hallmarks with genetic basis (as reiterated by the same authors in 2011 and by the voluminous treatise by Weinberg in 2014) [35- 37]. Historical and logical needs for such a synthesis under a genetic umbrella are also unquestionable and will probably become object of future or epistemological studies. But, with the clock ticking toward the EOR's inevitable discoveries, the distinguo's started appearing and are growing. Cancer maybe is not or not just genetic. The first objections became from the field of epigenetics (S. Baylin, P. Johnes) and cytogenetics (P. Duesberg, G. Hen) [38-40]. Obviously, cancer cells often display also epigenetic and chromosomal hallmarks. Although the 2011 and 2014 version of HoC include clear examples of chromosomal or epigenetic derangements in cancer cells, the proposed picture privileges genetic alterations, which eventually impinge into the machinery regulating epigenesis and epigenetic marks, chromosomal segregation and structures, etc. [36, 37].
3. Rationalize And Identify Some Priorities For Future Successful Interventions
Are we, therefore, asking the right question(s)? In recent months, a paper in Science by Tomasetti and Vogelstein has stressed this enigma to the limit by showing a randomness in cancer hazard (incidence) [41]. Needless to say, this has stimulated strong opposition from cancer research areas working on environmental carcinogenesis, an important field started from K. Yamagiwa almost 100 years ago [42]. The Science paper has been misunderstood quite often by mass-media, TV etc., as pointed out in the clear analysis of L. Luzzatto in NEJM a few months ago, to which I refer for further clarifications [43]. Still, the emerging question is the one of causality (or lack-of as per Tomasetti and Vogelstein). Specific causality is clearly denied, if we pretend to know with certainty what cancer is, what I called the engine of cancer (TEOC). If we are totally sure that TEOC is somatic mutations accrued during life-time (much more rarely by inheritance), then cancer can have a random component as Tomasetti and Vogelstein have clearly shown [41]. The real question becomes the nature of TEOCs. To rephrase a well-known quote: "DOES THE DEVIL PLAY DICE?" I have already indicated friends and foes of such theory, but the logic tells us that we should probably look better –as for the HoC paradigm- at TEOCs: their origins and their evolutionary mechanisms. As previously indicated [1], simple reading of today's literature suggests that more mechanisms than just somatic mutations are proposed, are suggested or are believed to be at the origin of TEOC: at least 9 additional are summarized and discussed [1]. Another consideration (only marginally discussed in [1] and which I am expanding elsewhere), is that according to HoC and consequently in the great majority of Targeted Gene Therapy (TGT) approaches, the postulated underlying mechanism is one of "oncogene addiction" [44-46]. However, oncogene-addiction has never been clearly defined, particularly for its ontogeny and the failure of most TGT may be also linked to ambiguity of such concept (or misconcept) [1]. Modelling in Cancer Research and Biology in general appears to be much more slow-moving than in other scientific arena's: think about nuclear physics or astrophysics for a comparison [47]. This phenomenon was also discussed by Leslie Orgel in Nature [48]. In our cancer genetic-paradigm today, the model maintained for over 15 years is strongly a-symmetrical. As suggested by Vogelstein and Fearon 19 years ago, the yinyang forces of Oncogenes and TSGs should be complemented upstream only by so-called Caretakers with a mechanism resembling that of TSGs, but earlier in ontogeny (see Figure 1A) [49].
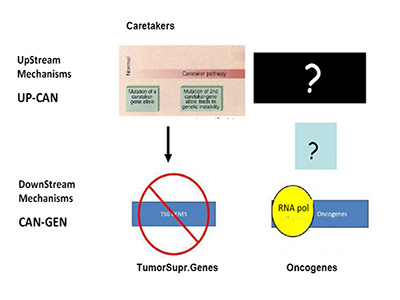
Figure 1A:Mechanism of Tumor Suppressor Genes and Oncogenes.
This mechanism is totally asymmetrical in proposing that upstream in cancer ontogeny only positive, beneficial, i.e. repair genes or Caretakers can be operating: this great logical gap is represented in Figure 1A as a Black upper right Box. However, as I have recently proposed, logics and experimental evidence suggest that also negative, pathological, genedeleterious mechanisms may be operating early in ontogeny, upstream not only of Oncogenes but also TSGs [5]. These genetic factors were called Genome-Snipers, to indicate the active nature of such mechanisms (see scheme in Figure 1B) [5].
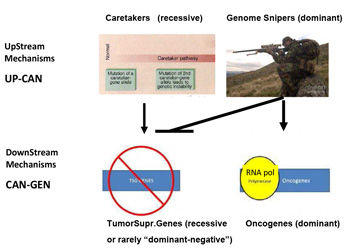
Figure 1b:Dominant mechanism of Genome Snipers on Tumor Suppressive Genes.
Older work and new evidence have pinpointed the potential relevance of such Genome-Sniper mechanisms, the main ones being:
- For several years, we have documented that infection with an RNA virus isolated from paediatric tumours (MicroFoci inducing Virus or MFV) induces sudden and dramatic oncogene amplification as well as genomic aberrations [51- 53].
- A phenomenon called chromothripsis or chromoplexis which creates sudden pulverization single or multiple chromosomes, with subsequent defective repair, has been described by several groups in bone, prostate and other cancers [54-58].
- Sottoriva et al. have recently proposed from NGS studies of colorectal cancer cells a similar mechanism of sudden and initial accrual of extensive mutations'very early in tumour ontogeny and not just as public or driver-, but also as private or passenger- mutations [50]. This was called the "Big Bang model of oncogenesis" [9].
CONCLUSION
The concluding remarks want to suggest that in an era in which NGS applications to cancer cells will become pervasive, it will be essential to also focus on data interpretation and not just on their accrual. Even the Hubble telescope would limit its analysis to a scanty number of galaxies, if fixed at a single angle of the universe. Today's analysis cannot be restricted to the idea that somatic mutations "must" be the only causes of human cancer, it has to become more comprehensive and should finally provide explicatory mechanisms for the plethora of additional phenomenology and models emerging in cancer research [1, 7, 59-63].
REFERENCES
- Rovigatti U. (2015). Cancer modelling in the NGS era - Part I: Emerging technology and initial modelling. Critical Reviews in Oncology/Hematology. 96(2), 274-307.
- Luthra R, Chen H, Roy-Chowdhuri S, Singh RR, et al. (2015). Next- Generation Sequencing in Clinical Molecular Diagnostics of Cancer: Advantages and Challenges. Cancers. 7(4), 2023- 2036.
- Gray P, Dunlop C and A. Elliott. (2015). Not All Next Generation Sequencing Diagnostics are Created Equal: Understanding the Nuances of Solid Tumor Assay Design for Somatic Mutation Detection. Cancers. 7(3), 1313-1332.
- Shaw V, Bullock K and Greenhalf W. (2016). Single-Nucleotide Polymorphism to Associate Cancer Risk, in Cancer Gene Profiling SE - 6, R. Grützmann and C.D.-.-.-.-.-. Pilarsky, Editors. 2016, Springer New York DA - 2016/01/01. 93-110 LA - English.
- Rovigatti U. (2014). Pitfalls and perspectives in cancer genomes NGS studies: implications for predictive, preventive and personalized medicine (PPPM). EPMA J. 5(Suppl 1), A30.
- Rovigatti U. (2013). Is There an Infectious Agent Behind Prostate Cancer?. Advances in Prostate Cancer, Wien (AU), INTECH Publisher.
- Gatenby R. (2012). Perspective: Finding cancer's first principles. Nature. 491(7425), S55.
- Kreso A and Dick JE. (2014). Evolution of the Cancer Stem Cell Model. Cell Stem Cell. 14(3), 275-291.
- Sottoriva A, Kang H, Ma Z, Graham TA, et al. (2015). A Big Bang model of human colorectal tumor growth. Nat Genet. 47(3), 209-216.
- Burghel GJ, Hurst CD, Watson CM, Chambers PA, et al. (2015). Towards a Next-Generation Sequencing Diagnostic Service for Tumour Genotyping: A Comparison of Panels and Platforms. BioMed Research International. 2015(2015), 6.
- Vanni I, Coco S, Truini A, Rusmini M, et al. (2015). Next-Generation Sequencing Workflow for NSCLC Critical Samples Using a Targeted Sequencing Approach by Ion Torrent PGMTM Platform. International Journal of Molecular Sciences. 16(12), 28765-28782.
- McGinn S, Bauer D, Brefort T, Dong L, et al. (2016). New technologies for DNA analysis - a review of the READNA Project. New Biotechnology. 33(3), 311-330.
- Hunt M, Silva ND, Otto TD, Parkhill J, et al. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biology. 16(1), 294.
- Goodwin S, Gurtowski J, Ethe-Sayers S, Deshpande P, et al. (2015). Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Research. 25. (11), 1750-1756.
- Eisenstein M. (2012). Oxford Nanopore announcement sets sequencing sector abuzz. Nat Biotech. 30(4), 295-296.
- Stoddart D, Heron AJ, Mikhailova E, Maglia G, et al. (2009). Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proceedings of the National Academy of Sciences. 106(19), 7702-7707.
- Yang Y, Liu R, Xie H, Hui Y, et al. (2013). Advances in Nanopore Sequencing Technology. Journal of Nanoscience and Nanotechnology. 13(7), 4521-4538.
- Butler TZ, Pavlenokb M, Derringtona IM, Niederweisb M, et al. (2008). Single-molecule DNA detection with an engineered MspA protein nanopore. Proceedings of the National Academy of Sciences. 105(52), 20647-20652.
- Manrao EA, Derrington IM, Pavlenok M, Niederweis M, Gundlach JH. (2011). Nucleotide Discrimination with DNA Immobilized in the MspA Nanopore. PLoS ONE. 6(10), e25723EP.
- Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, et al. (2014). Decoding long nanopore sequencing reads of natural DNA. Nat Biotech. 32(8), 829-833.
- Quick J, Quinlan AR and Loman NJ. (2014). A reference bacterial genome dataset generated on the MinION-TM portable single-molecule nanopore sequencer. GigaScience. 3(22).
- Loman NJ and Quinlan AR. (2014). Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics. 30(23), 3399-3401.
- Watson M, Thomson M, Risse J, Talbot R, et al. (2015). poRe: an R package for the visualization and analysis of nanopore sequencing data. Bioinformatics. 31(1), 114-115.
- Kielbasa SM, Wan R, Sato K, Horton P, Frith MC. (2011). Adaptive seeds tame genomic sequence comparison. Genome Research. 21(3), 487-493.
- Chaisson MJ and Tesler G. (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. Bioinformatics. 13(1), 1-18.
- Peng X, Wang J, Zhang Z, Xiao Q, et al. (2015). Realignment of the unmapped reads with base quality score. BMC Bioinformatics. 16(Suppl 5), S8.
- Jain M, Fiddes IT, Miga KH, Olsen HE, et al. (2015). Improved data analysis for the MinION nanopore sequencer. Nat Meth. 12(4), 351-356.
- minoTour, http://minotour.nottingham.ac.uk, 2015.
- Leggett RM, Heavens D, Caccamo M, Clark MD, et al. (2016). NanoOK: multi-reference alignment analysis of nanopore sequencing data, quality and error profiles. Bioinformatics. 32(1), 142-144.
- Church GM. (2006). Genomes for all. Sci Am.. 294(1), 46- 54.
- Judson HF. (1996). The Eighth Day of Creation: The Makers of the Revolution in Biology (Commemorative Edition). 1, CSHLP.
- Benzer S. (1955). Fine Structure of a Genetic Region in Bacteripophage. Proceedings of the National Academy of Sciences of the USA. 41(6), 344-354.
- Benzer S. (1959). On the topology of the genetic fine structure. Proceedings of the National Academy of Sciences of the USA. 45(11), 1607-1620.
- Shapiro J, Machattie L, Eron L, Ihler G, et al. (1969). Isolation of pure lac operon DNA. Nature. 224(5221), 768-74.
- Hanahan D and Weinberg RA. (2000). The hallmarks of cancer. Cell. 100(1), 57-70.
- Hanahan D and Weinberg RA. (2011). Hallmarks of Cancer: The Next Generation. Cell. 144(5), 646-674.
- Weinberg RA. (2014). The Biology of Cancer. 2nd Edition ed. 1, Garland Science.
- Baylin SB and Jones PA. (2011). A decade of exploring the cancer epigenom- biological and translational implications. Nature Rev Cancer. 11(10), 726-734.
- McCormack A, Fan JL, Duesberg M, Bloomfield M, et al. (2013). Individual karyotypes at the origins of cervical carcinomas. Molecular Cytogenetics. 6(1), 44.
- Heng H, Bremer SW, Stevens JB, Horne SD, et al. (2013). Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer and Metastasis Reviews. 32(3-4), 325-340.
- Tomasetti C and Vogelstein B. (2015). Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 347(6217), 78-81.
- Fujiki H. (2014). Gist of Dr. Katsusaburo Yamagiwa's papers entitled: "Experimental study on the pathogenesis of epithelial tumors" (I to VI reports). Cancer Science, 105(2), 143-149.
- Luzzatto L and Pandolfi PP. (2015). Causality and Chance in the Development of Cancer. New England Journal of Medicine. 373(1), 84-88.
- Lees JA and Weinberg RA. (1999). Tossing monkey wrenches into the clock: New ways of treating cancer. Proceedings of the National Academy of Sciences. 96(8), 4221-4223.
- Weinstein IB. (2002). Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 297(5578), 63-64.
- Gazdar AF, Shigematsu I, Herz J, Minna JD, et al. (2004). Mutations and addiction to EGFR: the Achilles "heal" of lung cancers? Trends in Molecular Medicine. 10(10), 481-486.
- Hawking, The Grand Design. 1st ed. 2010, New York: Bantam Books. 198.
- Orgel LE. (1999). Are you serious, Dr Mitchell? Nature. 402(6757), 17.
- Kinzler KW and Vogelstein B. (1997). Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 386(6627), 761- 763.
- Kang H, Salomon MP, Sottoriva A, Zhao J, et al. (2015). Many private mutations originate from the first few divisions of a human colorectal adenoma. The Journal of Pathology. 237(3), 355-362.
- Rovigatti U. (1992). Isolation and initial characterization of a new virus: Micro-Foci inducing virus or MFV. CR Acad Sci III. 315(5), 195-202.
- Rovigatti U, Piccin ATA, Colognato R and Sordat B. (2004). Preliminary Characterization of a New Type of Viruses Isolated from Paediatric Neuroblastoma and Non- Hodgkin's Lymphoma: potential Implications for Aetiology. in Intn. Conference Childhood Leukaemia. Section P1-18 I-IV, September. London, UK.
- Rovigatti U. (2012). Chronic Fatigue Syndrome (CFS) and Cancer Related Fatigue (CRF): two "fatigue" syndromes with overlapping symptoms and possibly related aetiologies. Neuromuscular Disorders. 22(2012), S235-S241.
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, et al. (2013). Punctuated Evolution of Prostate Cancer Genomes. Cell. 153(3), 666-677.
- Shen MM. (2013). Chromoplexy: A New Category of Complex Rearrangements in the Cancer Genome. Cancer Cell. 23(5), 567-569.
- Maher CA and Wilson RK. (2012). Chromothripsis and Human Disease: Piecing Together the Shattering Process. Cell. 148(1-2), 29-32.
- Forment JV, Kaidi A and Jackson SP. (2012). Chromothripsis and cancer: causes and consequences of chromosome shattering. Nature Rev Cancer. 12(10), 663-670.
- Kloosterman WP, Koster J and Molenaar JJ. (2014). Prevalence and clinical implications of chromothripsis in cancer genomes. Curr Opin Oncol. 26(1), 64-72.
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, et al. (2013). Cancer Genome Landscapes. Science. 339(6127), 1546 - 1558.
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, et al. (2013). Signatures of mutational processes in human cancer. Nature. 500(7463), 415-421.
- Greaves M and Maley CC. (2012). Clonal evolution in cancer. Nature. 481(7381), 306-313.
- Dick JE. (2008). Stem cell concepts renew cancer research Blood. 112 (13), 4793-4807.
- Couzin-Frankel J. (2013). Breakthrough of the year 2013. Cancer Immunotherapy Science. 342(6165), 1432-1433.