Information Links
Related Conferences
Previous Issues Volume 7, Issue 1 - 2022
Gastrointestinal Stromal Tumor (GIST) Cecal in Pediatrics? Clinical Case
César Abraham Ramírez-Franco1, Adrián Valle Partida2, Segundo Yépez-Vallejo3, Michelle Castro Barrios4, Davinia Suárez Flores5, Guillermo Padrón Arredondo6,*
1General Surgery Resident, General Surgery Service of Hospital General of Playa del Carmen, México
2Pediatric surgeon, General Surgery Service of Hospital General of Playa del Carmen, México
3,6General surgeons, General Surgery Service of Hospital General of Playa del Carmen, México
4,5Pathologists, General Surgery Service of Hospital General of Playa del Carmen, México
*Corresponding author: Guillermo Padrón-Arredondo, General Surgery Service of Hospital General of Playa del Carmen, Av. Constituyentes s/n with 35 Street, Col. Ejidal, Solidaridad Playa del Carmen, Q. Roo- PC: 77712, México, Tel: +52 984 876 2267, ORCID: 0000-0001-6049-5672; E-mail: [email protected].
Received Date: December 13, 2022
Published Date: December 31, 2022
Citation: Padrón-Arredondo G, et al. (2022). Gastrointestinal Stromal Tumor (GIST) Cecal in Pediatrics? Clinical Case. Mathews J Gastroenterol Hepatol. 7(1):17.
Copyrights: Padrón-Arredondo G, et al. © (2022).
ABSTRACT
Introduction: Gastrointestinal stromal tumors or GISTs are rare and occur mainly in the adult population, but a small percentage (1 to 2%) affect children and adolescents. GISTs are currently considered the most common mesenchymal neoplasm with a potential impact of around 1 to 1.5 cases per 100,000 inhabitants. Clinical Case: A 16-year-old female with no significant history of the case, began her condition with a month of evolution with postprandial fullness, occasional abdominal pain and hematuria. On physical examination it was globose at the expense of an intra-abdominal tumor of 40 x 50 cm of semisolid consistency, the rest of the examination without pathological data. Computed tomography reports “right ovarian tumor” measuring 21 x 22 x 14 cm. Normal laboratory studies. An exploratory laparotomy was performed, locating a tumor dependent on the cecum with implants in the adjacent peritoneum, an ectopic left kidney located in the left iliac fossa. Left hemicolectomy with side-to-side anastomosis is performed. Histopathological report reports spindle cell gastrointestinal stromal tumor with mild low-grade atypia (G1). Inflammatory (myoglandular) polyps with hyperplastic colonic mucosa florid with lymphoid follicles. Discussion: The best way to diagnose GISTs preoperatively is by fine-needle biopsy, although some fear that this system may spread tumors. Up to 85% of pediatric GISTs and 10-15% of adults do not contain the KIT or PDGFRA mutations and are classified as wild-type GISTs that are heterogeneous with respect to their clinical behavior and molecular profile.
Keywords: Gastrointestinal stromal tumors, Cecal, Pediatrics, CT.
INTRODUCTION
Gastrointestinal Stromal Tumors (GIST) are rare and occur primarily in the adult population, but a small percentage (1–2%) affects children and adolescents. GISTs are currently considered the most common mesenchymal neoplasm with a potential impact of around 1 to 1.5 cases per 100,000 inhabitants [1].
Childhood tumors are estimated to have an annual incidence of 0.02 per million children <14 years of age according to the UK National Registry. A recent review of the literature has described 21 familial cases (an association with neurofibromatosis type I or Carney's triad, with paraganglioma, lung chondroma, and esophageal leiomyoma) and 113 sporadic cases <21 years of age. Pediatric GIST: occurs mainly in women (70%), gastric (80%), and is often multifocal with typically slow development [1].
GISTs include benign lesions to malignant lesions whose origin may be in the digestive system, omentum, mesentery, and retroperitoneo; 50-60% appear in the stomach, 20-30% in the small intestine, 10% in the large intestine (rectum) and 55 in the esophagus [2,3]. The objective of the presentation of the case is to report a case of pediatric GIST of the cecal region in this hospital.
Around 85% of pediatric GISTs are wild-type (WT-GIST), which make up 10% of all diagnosed tumors of this type. They are often characterized by mutations or silencing of four genes encoding the subunits of the SDH enzyme complex. Moreover, the analyses show the frequent presence of NTRK/TRK (neurotrophic tyrosine kinase receptor) mutations (NRTK1, NTRK2, NTRK3), including fusions (e.g., ETV6:NTRK3, TPM3:NTRK1, TPR:NTRK1, LMNA:NTRK1, SPECC1L:NTRK3), in cases of WT-GISTs (5–25%). They can lead to different manifestations but also determine a specific treatment method. In parallel, they are characterized by the lack of canonical KIT, PDGFRA, SDHx, or RAS pathway components (KRAS, BRAF, NF1) alterations, which eliminates the possibility of the use of a range of multikinase inhibitors. Therefore, it is necessary to individualize treatment and apply other types of kinase inhibitors [4,5].
CLINICAL CASE
A 16-year-old female with no significant history of the case, began her condition with a month of evolution with postprandial fullness, occasional abdominal pain, and hematuria. On physical examination, the abdomen was globose at the expense of an intra-abdominal tumor of 40 x 50 cm with a semisolid consistency, the rest of the examination showed no pathological data. Computed tomography reports a right ovarian tumor measuring 21 x 22 x 14 cm. Normal laboratory studies. An exploratory laparotomy was performed, locating a tumor depends on the cecum (Figure 1) with implants in the adjacent peritoneum, and ectopic left kidney located in the left iliac fossa. Right hemicolectomy with side-to-side anastomosis is performed. Histopathology reports spindle cell gastrointestinal stromal tumor with mild low-grade atypia (G1) (Figure 2). Inflammatory (myoglandular) polyps with hyperplastic colonic mucosa florid with lymphoid follicles. Slides were sent to two cancer centers in Guadalajara and Campeche, México for immunohistochemical analysis without positive results for GIST but ruling out leiomyoma, schwannoma, and leiomyosarcoma.
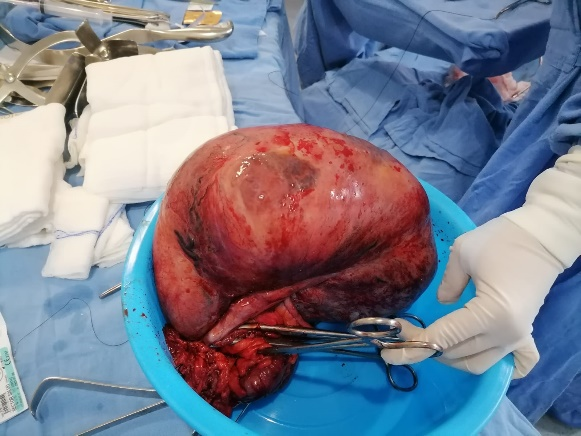
Figure 1. Tumor mass dependent on the cecum with a healthy appendix.
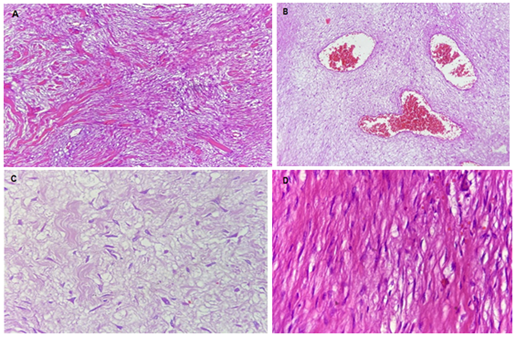
Figure 2. Photomicrographs of the tumor mass (GIST?).
Panoramic view (4x) of a neoplasm of mesenchymal origin characterized by intersecting fascicles, which may present variable hyalinization (A and edema B). C. At higher magnification (40x), thickened spindle cells, with mild to moderate atypia, is seen in an edematous stroma. D. Some of the neoplastic cells are more like a smooth muscle (40x), in a more hyalinized stroma.
DISCUSSION
Li C, et al. [6] carried out a comparative study between single and multiple GISTs (MGIST) and found that both have the same characteristics such as low morbidity, higher frequency in women, young age and involvement of multiple organs and although the total survival of both tumors is the same, the higher frequency of MGIST metastases affects the survival of patients, a case similar to ours.
Bulusu VR [7], reviewed 60 patients, 20 men; 40 women aged 38 years (range 14-76). Primary tumor location: 67% gastric and 33% small intestine. 10 GISTs were observed in patients with neurofibromatosis-1 (NF-1), located mainly in the small intestine and frequently multifocal. Twenty patients were treated with tyrosine kinase inhibitors. No objective responses were observed with imatinib. Disease stabilization was seen more frequently with regorafenib. Regarding the location of the tumor, the cecal location of our case is striking, which is very rare and we did not find anything similar in the bibliography.
GISTs smaller than 5 cm are considered benign and with little recurrence. In 95% of cases, it is necessary to determine the KIT [familial GIST with germline mutation] (CD117) which is a tyrosine kinase receptor on the interstitial cells of Cajal. Imaging studies in large tumors are tomography and magnetic resonance imaging with and without contrast medium.
Small tumors present as submucosal or endoluminal polyps. GISTs must be differentiated from leiomyomas, schwannoma, and leiomyosarcoma, although GISTs are hypervascular in the arterial phase when compared to adjacent tissues in the portal venous phase. Its most accepted management is block [8] resection as was performed with our patient.
The best way to diagnose GISTs preoperatively is by fine needle biopsy (FNA) although some fear that this system is a tumor spreader. Jakov J, et al. [9] carried out a systematic review to assess tumor recurrence due to BAAF biopsy without finding evidence of recurrence of the disease after the procedure (although one case did present GIST cell seeding), but they ensure that the method is useful for tumor differentiation and the corresponding treatment. They performed a systematic literature search and included studies assessing the oncological outcome of GIST patients who underwent a pre-treatment core needle biopsy or fine needle aspiration. Three non-randomized studies and eight case reports comprising 350 patients were eligible for inclusion. No prospective study designed to answer the review question was found. One case of needle tract seeding after percutaneous core needle biopsy of GIST was reported. None of the studies reported an increased rate of abdominal recurrence in patients with pretreatment biopsy. The existing evidence does not indicate a relevant risk of needle tract seeding or abdominal recurrence after pre-treatment biopsy of GIST. Biopsy can safely be done to differentiate GIST from other tumors and to select the most appropriate treatment.
GISTs are clinically detected by endoscopy, imaging studies, and immunostaining and require minimally invasive surgery; recently, neoadjuvant treatment has been applied when the tumor is large or has infiltrated adjacent organs. The combination with tyrosine kinase inhibitors is important to maximize the therapeutic effect of surgery. Imatinib is the standard treatment in the postoperative period. However, the effect of the tyrosine kinase inhibitor can vary in the mutation of the c-KIT genes at the site of the mutation, in addition, monitoring of possible recurrence is important, so its management must be multidisciplinary [10].
GISTs are sensitive to KIT/PDGFRA (gain-of-function mutations) inhibitors of tyrosine kinase (TKI). Imatinib is the first-line treatment in advanced metastatic GISTs, but imatinib alone does not eradicate GISTs and more than 90% of cases are resistant, and although third-generation tyrosine kinase has already been developed and is in clinical use, it continues to present resistance due to the appearance of clones with resistant mutations [11].
Up to 85% of GISTs in pediatrics and 10-15% of adults do not contain the KIT or PDGFRA mutations and are classified as wild-type GISTs (wt-GISTs) that are heterogeneous with respect to their clinical behavior and molecular profile, and they have been classified into a subclass as syndromic and non-syndromic or sporadic. Lately, the use of succinate dehydrogenase B (SDHB) has been used to stratify GIST into SDHB with retention and SDHB without retention or deficient, useful for predicting genetic changes and prognosis for this rare disease [12].
Singh AS et al. [13] in the study randomized phase II con nomolumab monoterapy vs. nivolumab plus ipilimumab in Gist patients enrolled 36 patients with advanced/metastatic GIST refractory to at least imatinib were randomized 1:1 in a non-comparative, parallel group, unblinded phase II trial of N (240 mg every 2 weeks) or N + I (240 mg every 2 weeks + 1 mg/kg every 6 weeks). The primary endpoint was the objective response rate of N alone or N+I by RECIST 1.1 in the intent-to-treat population. The primary endpoint of response rate > 15% was not observed for N or N + I. In a heavily pretreated GIST population, responses and long-term disease control with both N and N+I were observed. No new safety signals have been observed.
On the other hand, George S, et al. [14] in phase II study about ponatinib en advanced Gist Forty-five patients were enrolled (30 KIT ex11–positive and 15 KIT ex11–negative); median follow-up was 14.7 and 13.6 months, respectively, as of August 1, 2016. Sixteen-week CBR was 36% (KIT ex11–positive; primary endpoint) and 20% (KIT ex11–negative). ctDNA analyses (n = 37) demonstrated strong concordance of primary KIT mutations between plasma and tumor. At least two secondary mutations were detected in 35% of patients overall and 54% of KIT ex11–positive patients. Changes from baseline in mutated ctDNA levels were consistent with clinical activity. Ponatinib was ineffective in patients with KIT exon 9 primary mutations. Resistance was associated with the emergence of V654A. AOEs and venous thromboembolic events occurred in three and two patients, respectively. Six patients died; two deaths (pneumonia and pulmonary embolism) were considered possibly ponatinib-related. Thus, ponatinib demonstrated activity in advanced GIST, particularly in KIT ex11–positive disease. ctDNA analysis confirmed heterogeneous resistance mutations in TKI-pretreated advanced GIST. Safety was consistent with previous studies.
CONCLUSIONS
In this case, the clinical diagnoses is done but the histopathological diagnoses was very difficult because we answer different pathologists from three hospitals of oncology specialties with the actual technology and they cannot establish the final diagnostics.
ETHICAL CONFLICTS
None.
CONFLICT OF INTERESTS
None.
FINANCIAL SUPPORT
None.
REFERENCES
- Morales PA, Millán VLO, Covarrubias EG, et al. (2017). Pediatric GIST tumor. Presentation of two cases and review of the literature. Bol Clin Hosp Infant Edo Son. 34(2):127-135.
- Verdecia-Cañizares C, Villamil-Martínez R, Montero-Reyes I, Pineda-Fernández D. (2017). Gastrointestinal stromal tumor. Case presentation. Rev Cub Ped. 89(1):53-59.
- Shimomura M, Ikeda S, Takakura Y, Kawaguchi Y, Tokunaga M, Takeda H, et al. (2010). Gastrointestinal stromal tumors of the small intestine in pediatric populations: a case report and literature review. Pediatric Surg Int. 26(6):649-654. DOI: 10.1007/s00383-010-2596-3.
- Andrzejewska M, Czarny J, Derwich K. (2022). Latest Advances in the Management of Pediatric Gastrointestinal Stromal Tumors. Cancers. 14(20):4989. DOI: 10.3390/cancers14204989.
- Casali PG, Abecassis N, Aro HT, Bauer S, Biagini R, Bielack S, et al. (2018). Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 29(Suppl 4):iv68-iv78.
- Li C, Yang KL, Wang Q, Tian JH, Li Y, Gao ZD, et al. (2020). Clinical features of multiple gastrointestinal stromal tumors: A pooling analysis combined with evidence and gap map. World J Gastroenterol. 26(47):7550-7567. DOI: 10.3748/wjg.v26.i47.7550.
- Bulusu VR, Casey R, Giger O, Maher E, Hardwick R, Carroll N, et al. (2018). Pediatric, adolescent, wild type, syndromic gastrointestinal stromal tumors (PAWS-GIST): Report from United Kingdom PAWS-GIST clinic. J Clin Oncol. 36(Suppl 15):e23519-e23519.
- Özcan HN , Yavuz ÖÖ, Ekinci S, Oguz B, Mithat Haliglu TK. (2022). Imaging findings of gastrointestinal tract tumors in children and adolescents. Insights into Image. 13:51. DOI: 10.1186/s13244-022-01193-9.
- Jakob J, Salameh R, Wichmann D, Charalambous N, Zygmunt A‑Ch, Kreisel I, et al. (2022). Needle tract seeding and abdominal recurrence following pre‑treatment Biopsy of gastrointestinal stromal tumors (GIST): results of a systematic review. BMC Surg. 22:202. DOI: DOI: 10.1186/s12893-022-01648-2.
- Sugiyama Y, Sasaki M, Kouyama M, Tazaki T, Takahashi S, Nakamitsu A. (2022). Current treatment strategies and future perspectives for gastrointestinal stromal tumors. World J Gastrointest Pathophysiol. 13(1):15-33.
- Hayashi Y, Vy Nguyen TT. (2021). A narrative review of imatinib-resistant gastrointestinal stromal tumors. Gastrointest Stromal Tumor. 4:6. DOI: 10.21037/gist-21-10.
- Brčić I, Argyropoulos A, Liegl-Atzwanger B. (2021). Update on Molecular Genetics of Gastrointestinal Stromal Tumors. Diagnostic. 11(2):194. DOI: 10.3390/diagnostics11020194.
- Singh AS, Hecht JR, Rosen L, Wainberg ZA, Wang X, Douek M, et al. (2022). A Randomized Phase II Study of Nivolumab Monotherapy or Nivolumab Combined with Ipilimumab in Patients with Advanced Gastrointestinal Stromal Tumors. Clin Cancer Res. 28(1):84-94.
- George S, von Mehren M, Fletcher JA, Sun J, Zhang S, Pritchard JR, et al. (2022). Phase II Study of Ponatinib in Advanced Gastrointestinal Stromal Tumors: Efficacy, Safety, and Impact of Liquid Biopsy and Other Biomarkers. Clinical Cancer Res. 28(7):1268-1276.