Previous Issues Volume 3, Issue 1 - 2018
CCl4 Induced Liver Fibrosis Model in Cynomolgus Monkeys
Shanshan Ding*1 , Changqing Zhang1 , Li Zhang2 , Lei Le1 , Xin Zhang3 , Joyce Hwa2 , Tiansheng Zhou4 , Shoutao Liu1 , Zhihai Li1
1 DMPK-PD department, WuXi AppTec, Wuzhong District, Suzhou, Jiangsu Province, China.
2 WuXi AppTec / XenoBiotic Laboratories, 107 Morgan Lane, Plainsboro, New Jersey 08536.
3 DMPK-PD department, WuXi AppTec, 288 Fute Zhong Road, Waigaoqiao Free Trade Zone, Shanghai, China.
4 Pathology department, WuXi AppTec, Wuzhong District, Suzhou, Jiangsu Province, China.
Corresponding Author: Shanshan Ding, WuXi AppTec, Wuzhong District, Suzhou, Jiangsu Province, China, Tel: + 8651266374713; Email: [email protected]
Received Date: 22 May 2018 Accepted Date: 20 Jun 2018 Published Date: 27 Jun 2018
Copyright © 2018 Ding S
Citation: Ding S, Zhang C, Zhang L, Le L, et all. (2018). CCl4 Induced Liver Fibrosis Model in Cynomolgus Monkeys. M J Vetr. 3(1): 009. Citation: Ding S, Zhang C, Zhang L, Le L, et all. (2018). CCl4 Induced Liver Fibrosis Model in Cynomolgus Monkeys. Mathews J Vet Sci. 3(1): 009.
ABSTRACT
Liver fibrosis is the final common stage of the most chronic liver diseases, it is caused by several factors which lead to a major worldwide health care burden. Over the decades, the understanding of the liver fibrosis disease was growing rapidly, several studies reported that this progress could be regressed or reversed, which give us a bright prospect in developing anti-fibrotic therapies. In this experiment, liver fibrosis was fully developed after CCl4 induction for 7 weeks in eight animals. Clinical pathologic parameters, four indicators of hepatic fibrosis in monkey showed similarly changes in human. All animals had liver fibrosis after 1.5 months of CCl4 induction, and liver fibrosis still existed after 9 months recovery periods, the fibrosis stages in most animals had no obvious regression without treatment.
Keywords: Liver Fibrosis; Animal Model; CCl4; Reverse.
INTRODUCTION
Liver fibrosis is defined as an abnormal response of the liver to persistent injury, characterized by the excessive accumulation of collagenous extracellular matrices (ECMs), and therefore involves both wound healing and fibrotic processes [1-3]. The repair processes occurs right after liver injury, which can take either of two distinct paths: one way called regenerative path in which injured cells are replaced by the same type of cells; the other is connective tissue replaces normal parenchymal tissue in an uncontrolled fashion, which is known as fibroplasias or fibrosis [4-8]. Persisting injury caused uncontrolled repair processes, lead to the damaged tissues/organs undergo substitution by over-abundant ECM and suffer from extensive, pathological fibrosis [3]. The onset of liver fibrosis is usually insidious, advanced liver fibrosis results in liver failure and portal hypertension and is associated with an increased risk of liver cancer [9]. Severe end-stage liver disease (cirrhosis or hepatoceliular carcinoma) is associated with morbidity and mortality, and orthotopic liver transplantation is often indicated as the only effective therapy [10]. However, liver transplantation has several disadvantages, shortages of organ donors, the commitment of recipients to lifelong toxic immunosuppression, and recrudescence of the original disease in transplant recipients, therefore effective antifibrotic treatments are urgent unmet medical needs [11, 12]. Liver fibrosis research can be assigned to two broad groups: in-vitro model including cell culture model [13, 14] human tissue culture [15], and in-vivo experimental animal models. Celbehavior and the effect of specific mediator could be studied in in-vitro model, but it clearly cannot recapitulate the event that occur in-vivo. As we all know, liver fibrosis is developing disease with potentially dynamic processes that resulted from the complexed interplay of resident and incoming cells in a microenvironment. Animal models have been used for several decades to study fibrogenesis and to validate anti-fibrotic effects of potential therapeutic approaches [16, 17]. Animal models allow for (i) comprehensive study of questions that may not be able to address in human studies, (ii) multiple sampling at strategic times during the development vs. resolution phases, (iii) experimental testing with restriction of the minimal number of variables [18].
Current animal model in liver fibrosis research are allocated in four main categories, the first category is via the cholestatic mechanism that damage the biliary epithelium including surgical bile duct ligation model [19], gene knockout or transgenic model [20, 21], dietary models by feeding with 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) or α-naphthylisothiocyanate (ANIT) [22, 23]. The second category is induced by hepatotoxins such as CCl4 [24], thioacetamide (TAA) [25], or dimethylnitrosamine (DMN) [26] that belong into toxin-induced liver models. The third category is activated by metabolic liver injuries including both alcoholinduced fibrosis and NASH-associated fibrosis [27-30]. The fourth category is induced by autoimmune responses via injecting heterologous serum to elicit liver fibrosis [31]. Most of these models were established in rodent animals. Although rodent models can mimic the liver fibrosis development to some extent, several differences between murine and human need to take into consideration; such as the different number and proportion of distinct immune cell populations in the liver and the different marker molecules to identify corresponding immune cell subsets [32], and diversity in RNA expression is reflecting the fundamental physiological differences between mice and humans [33]. Studies revealed that the subsets of circulating classical and non-classical monocytes show very different ratios in humans (90%:10%) and mice (50%:50%) [34]. Nonhuman primates are essential and irreplaceable animal models in human disease research because genetic, anatomical and physiological similarity to humans.
High-fat diet and/or CCl4 induced rodent liver fibrosis was widely investigated [24, 35], but few studies report monkey liver fibrosis. Alcohol induced liver fibrosis model were developed in rhesus monkeys, which take 3 years [36]. Another study combined CCl4 subcutaneous dosing with chronically fed high-fat diet and alcohol in drinking water for 16 weeks to establish liver fibrosis model in cynomolgus monkeys [37]. Both studies used alcohol as a major inducer. In order to establish a non-alcoholic liver fibrosis monkey model with a single stimulus within a reasonable time frame and to selectively target the liver, we chose to deliver CCl4 through the portal vein.
MATERIAL AND METHOD
Animal and Husbandry
Cynomolgus monkeys (3-6 years, 3-7 kg) were provided by Hainan Jingang Biotech Co., Ltd. All animals were singlehoused in stainless steel cages equipped with a bar type floor and an automatic watering valve, these cages conform to standards set forth by the US Animal Welfare Act. The rooms controlled humidity at 40% to 70%, temperature at 18°C to 29°C, 10 to 20 air changes/hour and 12-hour light/dark. Regular or high fat diet and fresh fruit were fed daily. Protocols for all the animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) (WuXiAppTec Co., Ltd, Suzhou, Jiangsu province, The People’s Republic of China).
Reagent and Food
Analytical Grade reagent CCl4 (catalog no. 20050521, Sinopharm Chemical Reagent Co.,Ltd), PEG 400 (catalog no. MKBG7718V). Ketamine hydrochloride (catalog no. 1507293, Fujian Gutian Pharma Co., Ltd.).
Experiment
Animals had portal vein cannulation surgery. Briefly, animals were anesthetized through trachea intubation with isoflurane during surgery, the animals lied on its back and general sterilized in operation area, exposed portal vein and selected a branch of mesenteric vein at the far end. PE catheter was cannulated into the portal vein. After securing the catheter, the other end of catheter was connected with a heparin cap to confirm the catheter unobstructed. The heparin cap were placed in muscle layer subcutaneously. After a 20- 28 days recovery period, the animals were ready to use.
Eight convalescent portal vein cannulated animals were assigned into this experiment. Animals were dosed with CCl4 formulated in PEG 400 (400 mL/L) via intravenous bolus injection into portal vein. Animals were received escalating dosage at 0.1 mL/kg once weekly, 0.1 mL/kg twice weekly and 0.15 mL/kg twice weekly (Figure 1), all animals were put into recovery phase after the last dose.
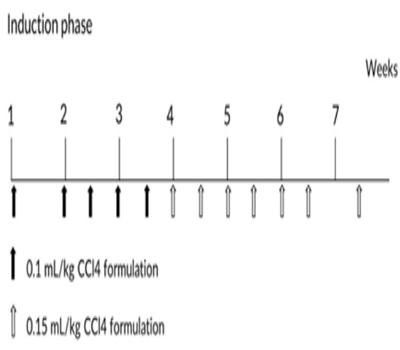
Figure 1Dose schedule of CCl4 during model induction phase.
Blood samples were collected before and weeks 1, 2, 4, 6, 8, 12, 24, 46 after first dosing, all blood samples were collected from a peripheral vessel into commercially available tubes containing Potassium (K2 ) EDTA or plain with separating gel on the specified day. Serum samples were stored at -60o C or lower until analysis.
Liver biopsy and ultrasound B examination were conducted in this experiment. Animals were anesthetized with ketamine hydrochloride (10 mg/kg), lied on his back, sterilized appropriately, used ultrasound B (Vet-M7, Mindray) to keep away from big vessel and gall bladder, and then inserted auto biopsy gun (acecut 14G x 115mm, TSK, Japan) to collect liver tissue. After the procedure, animals were observed daily by experienced technician till its recovery.
Sample analysis
Whole blood samples (anti-coagulation EDTAK2 ) for hematological parameters were analyzed by an automatic analyzer (ADVIA 2120, Siemens). Serum samples for clinical chemistry parameters were detected by an automatic analyzer (HITACHI 7180, Hitachi High-Tech Science Systems Corporation). Serum samples for four indicators of hepatic fibrosis laminin (LN), hyaluronic acid (HA), collagen type IV (CIV), and N-terminal propeptide of collagen III (PIIINP) parameters were determined through radio immunoassay (RIA) method in ADC CLIA 400 automatic plate immunoassay analyzer (Autobio).
Pathological examinations
Liver tissue or biopsy samples were fixed in 10% formaldehyde, trimmed, processed, embedded in paraffin, sectioned, stained with hematoxylin and eosin and sirius red staining, and then examined microscopically. Liver fibrosis is classified by using Metavir system [38]: No fibrosis (F0), Fibrous portal expansion (F1), Few bridges or septa (F2), Numerous bridges or septa (F3) and Cirrhosis (F4)
RESULTS
Monkeys were dosed for up to 7 weeks, total CCl4 dose volume was from 1.43 to 3.46 mL. All animals entered into recovery phase after last dosing. The mean animal body weight (4.61±0.56 kg) decreased about 9% (4.20 ±0.48 kg) on week 7, but increased to 4.82±0.42 kg and 5.45±0.52 kg at 6 and 12 months respectively (Figure 2).
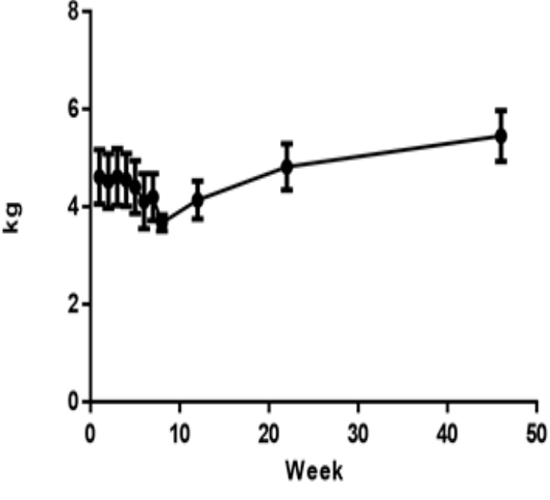
Figure 2 :Animal body weight changes in this study (n=8). Values are expressed as the mean ±SEM.
Liver enzymes Aspartic Transaminase (AST), Alanine Aminotransferase (ALT), Alkaline Phosphatase (ALP), GammaglutamylTranspeptidase (GGT) concentration were increased significantly after CCl4 induction, the mean peak levels were 77.6±9.37 U/L, 1071±146 U/L, 1482±453 U/L and 151±29.3 U/L respectively. (Figure 3). Total Bilirubin (TBIL) level was increased and reached to peak (8.4±1.64 µmol/L) at week 4. The total protein (TP), albumin (ALB) and albumin/globulin (A/G) ratio were declined 11% (70.2±1.98 g/L), 25% (31.2±1.26 g/L) and 41% (0.69±0.11) after dosing of CCl4 (Figure 4). All changed values returned gradually to normal in recovery period. Other clinical chemistry parameters do not change significantly. Whole hematology parameters including red blood cell, white blood cell, hemoglobin and other related items were in normal range during this experiment (data not shown).
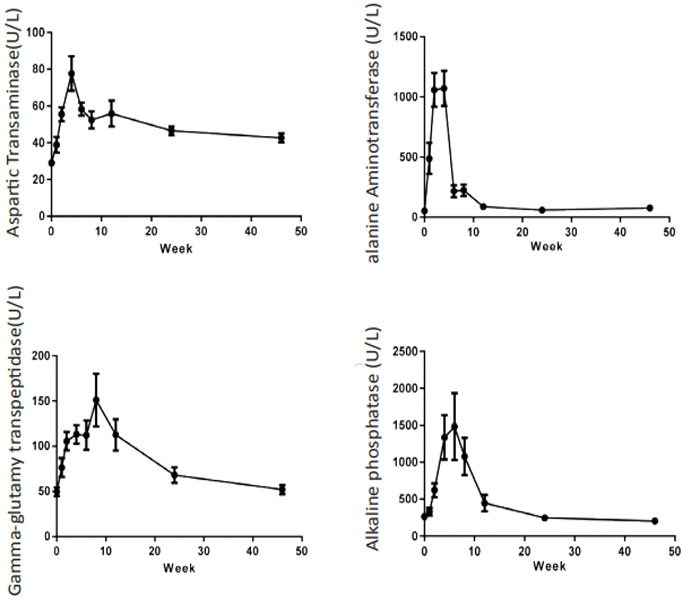
Figure 3: Sequential changes of liver enzymes in the process of liver fibrosis (n=8). Values are expressed as the mean ± SEM.
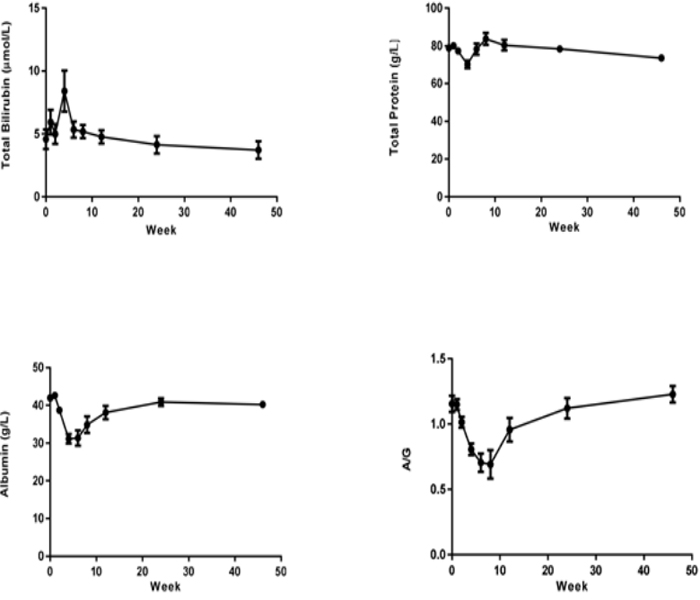
Figure 4: Sequential changes of other clinical pathologic parameters in the process of liver fibrosis (n=8). Values are expressed as the mean shown).
The HA, LN, and PIIINP parameters were increased from 72.8±21.6 ng/mL to 136±32.0 ng/mL, 201± 16.9ng/mL to 299±28.8 ng/mL, 26.1±5.27 ng/mL to 49.5±5.94 ng/mL after CCl4 induction respectively. HA and LN level restored to normal after a recovery periods, but the PIIINP value was still higher at week 24 than baseline (Figure 5). The mean CIV value was 34 ng/mL in week 4, beside that all the other CIV values were below the limit of quantification (15 ng/mL).
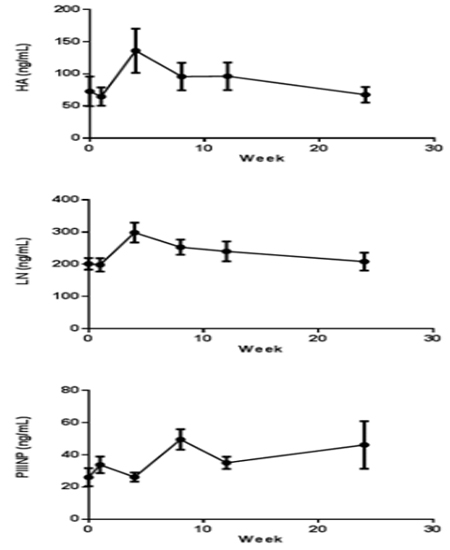
Figure 5: Indicators of hepatic fibrosis curve in cymonolgus monkeys pre and post CCl4 induction (n=8). Values are expressed as the mean ±SEM.
Pathology examination in liver biopsy samples showed that fibrosis was found for all animals (Figure 6). Liver fibrosis were existed persistently during the recovery period (Table 2), it did not cure naturally without treatment. Irregular or nodular surface and blunt edges in liver were observed under ultrasound B examination (Figure 7).
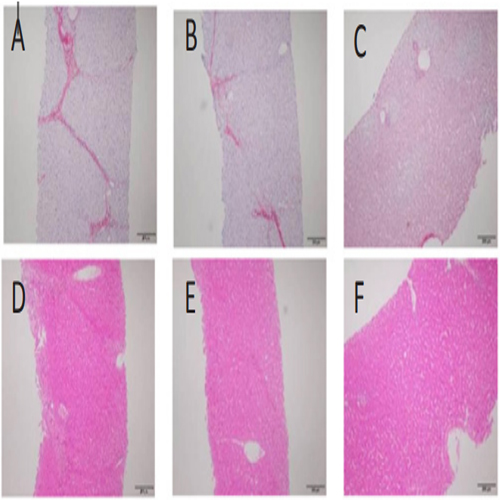
Figure 6: Pathological changes in liver tissue (200 X). The pictures sirius red staining (A) and HE staining (D) are presented F3, which the formed numerous bridges or septa, small number of pigmented macrophages (hemosiderin) and mononuclear inflammatory cells were observed. The pictures (B, E) are presented F2, few bridges or septa with inflammatory cells. And the pictures (C, F) are normal liver.
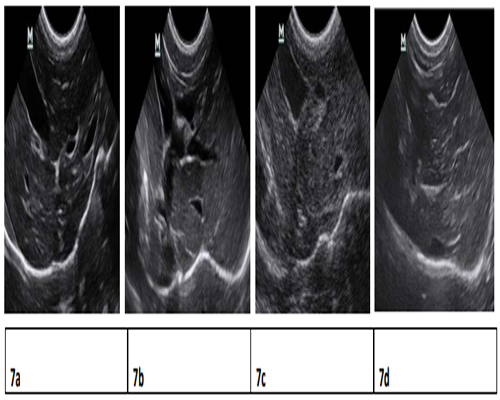
Figure 7:Ultrasound Liver images before induction, 1.5 months, 3 months, 11 months after induction. 7a) clear liver edge, smooth envelope, uniform echo from liver parenchyna, the structure and track of vessels are normal. 7b) obtuse and thick liver edge, parenchyma echo coarsened, increased liver volume and expansive portal vein. 7c) enchanced punctiform echo in parenchyma, rough liver edge, the branch of portal vein is a bate and the vein is abate and the vein wall is blur. 7d) strong echo structure in parenchyma, thickening liver edge.
Table 1: Suspected adverse reactions for small animals (dogs, cats) vaccines reported to the Canadian Centre for Veterinary Biologics between 2010 and 2014.
| Stage | Histologic description |
|---|---|
| 0 | No fibrosis |
| 1 | Zone 3 perisinusoidal fibrosis only |
| 2 | Zone 3 plus portal/periportal fibrosis |
| 3 | As above with bridging fibrosis |
| 4 | Cirrhosis |
Adapted from Brunt et al [1].
Table 2: Liver fibrosis stages for individual animal at different months after initial CCl4 dosing.
| Animal | 1.5 months | 3 months | 6 months | 11 months |
|---|---|---|---|---|
| 1 | 1 | 2 | 2 | 1 |
| 2 | 3 | 3 | 3 | 2 |
| 3 | 3 | 2 | 2 | 2 |
| 4 | 3 | 4 | 4 | 3 |
| 5 | 2 | 2 | 2 | 2 |
| 6 | 2 | 3 | 3 | 3 |
| 7 | 2 | 1 | 2 | 3 |
| 8 | 2 | 2 | 2 | 2 |
DISCUSSIONS
The kinetics of fibrosis development can be roughly divided into three phases: acute injury, initiation of fiber formation and advanced fibrosis [39]. CCl4 is metabolized by hepatocytes, giving rise to toxic trichloromethyl (CCl3) radicals by CYP2E1, an enzyme expressed in perivenular hepatocytes. It induces thus an acute centrolobular necrosis which triggers a wound healing response: 1. recruitment of phagocytic and inflammatory cells to clear necrotic zones, 2. activation of fibrogenesis and increased ECM, 3. proliferation of parenchymal and non-parenchymal cells to replace dead cells; which would restitute liver integrity. When the insult is repeated, successive rounds of wound healing occur prior to resolution of the previous one resulting in fibrosis accumulation [18]. All animals developed liver fibrosis after CCl4 administration via portal vein. Hemolysis could be induced rapidly when CCl4 quickly injected into portal vein, and liver cell necrosis could reduce the liver’s ability to metabolize and excrete bilirubin leading to a buildup of unconjugated bilirubin in the blood.
Liver fibrosis evaluation methods can be divided into invasive and non-invasive [40]. Non-invasive methods include serum tests, RNA expression analysis and imaging techniques. These methods may be performed repeatedly, allowing for ongoing monitoring of potential fibrosis in vivo [41]. In this study, the mean ALT was increased almost 20-fold after administrating CCl4. ALT was released from liver tissue into the circulation in proportion to the degree of hepatocellular damage. Its level is thought to be one of the most sensitive markers of liver injury and liver disease progression [42]. Mean AST level increased less than 3-fold after CCl4 induction. ALT is predominantly found in the liver, with clinically negligible quantities found in the kidneys, heart, and skeletal muscle. In contrast, AST is found in the liver, heart (cardiac muscle), skeletal muscle, kidneys, brain, and red blood cells. Therefore, ALT is a more specific indicator of liver damage than AST. The increasing of four liver enzymes AST, ALT, ALP, GGT levels and TBIL indicate liver toxicity.
ALB and TP, and A/G ratio were decreased. ALB is produced in the liver, impaired liver cannot synthesized effectively and maintain ALB level, whereas, globulins are produced in the liver or immune system. This might be the reason why GLB is not changed during CCL4 induction. The ratio of AST/ALT˃1 (AAR) has been proposed as a test of cirrhosis in human [43], while other study demonstrate that AST/ALT ratio is confounded when used in alcoholic and many other acute and chronic fatty infiltrating liver diseases [44], and not recommended for evaluation the stage of fibrosis. Among the monkeys diagnosed as liver fibrosis, the AST/ALT ratios were below 1.0 throughout the study.
The process of liver fibrosis is characterized mainly by cellular activation of hepatic stellate cells (HSCs) and are able to express and deposit large quantities of extracellular matrix components [45, 46]. Liver ECM components include collagen type I, III, and IV, fibronectin, undulin, elastin, laminin, hyaluronan, and proteoglycans were higher than normal in advanced stage [47]. HA, LN, PIIINP were increased, those were consistent with previous studies [48-50]. But N-terminal pro-peptide of collagen type III (PIIINP) level also elevated in chronic pancreatitis [44] and HA levels may be elevated after meal or glucose drink [51], they are not specific for liver fibrosis.
The ideal biomarker should: 1) Specific for liver; 2) Readily available and standardized between all laboratories performing diagnostic biochemistry/haematology; 3) Not subject to false positive results, for example due to inflammation; 4) Identifies the stage of fibrosis [52]. Currently, no non-invasive markers are specific and capable of providing accurate information about fibrogenesis and the extent of fibrosis in the liver. The utility of serum models such as Fibrotest [53], Fibrometer [54], Fibrospect [55], Hepascore [56] were used to predict fibrogenesis, but currently cannot replace the gold-standard method liver biopsy [57].
Fibrosis stage is assessed by Metavir (stage 0-4) score. We can found that increased fibrillar eosinophilic material (H&E stained slides) and red Sirius Red stained were noted in the periportal (centroacinar) area, this change generally limited to individual lobules, but also with extension from one portal tract to another (bridging fibrosis), in addition, small number of pigmented macrophages (hemosiderin) and mononuclear inflammatory cells were present.
However, there were some limitations when using liver biopsy evaluation. Firstly, hepatic fibrosis may not be homogenous throughout the liver, the size of biopsy specimen is not large enough to contain whole hepatic lobule, and it only represents a tiny fraction of organ. Sampling error (25%-40%) may result in poor reproducibility [58]. Secondly, it’s an invasive procedure that caused pain and major complication occurring in 40% and 0.5% of patients, respectively [59]. Thirdly, there is well known observer variability amongst pathologists in categorizing the degree of fibrosis, no matter how precisely defined the stage [60]. The liver fibrosis scores minor changed in different months in our experiment, it mainly depend on the liver biopsy sample size and sampling location, some histopathologic images including whole hepatic lobules which contribute to make judgement, and it’s really challenge to evaluate the fibrosis score in images with partial hepatic lobule. Increasing the biopsy sample numbers may decrease the erroneous judgement, but noting that biopsy is an invasive procedureMany imaging techniques have emerged for liver fibrosis detection and assessment, such as ultrasound [61], computed tomography (CT) [62] and magnetic resonance imaging (MRI) [63]. The image of ultrasound B showed clearly changes during the induction in our study, but it only produce specific findings, with very limited sensitivity and cannot assess the fibrosis stage, especially in early and intermediate stages. CT and MRI have the same problem [64, 65]. All in all, it would be better to combine both non-invasive and invasive method for comprehensive assessment of the liver stage
Liver fibrosis reversal is still a debated topic. When administrating of neutralizing TIMP1-specific antibody decreases the collagen content in CCl4-induced fibrosis [53], and the reversibility of fibrosis was found in experimentally induced cholestasis in rat [56]. In humans, spontaneous resolution of liver fibrosis can occur after successful treatment of the underlying disease. Hepatitis C caused liver fibrosis could be reverse after treatment [54]. It may take years for significant regression to be achieved, the time course varies depending on the underlying cause of the liver disease and its severity. Some experimental evidence suggests cirrhosis might reach a point of no return. Using the CCl4-intoxication rat model of liver fibrosis, the remodeling of advanced cirrhosis is limited and the liver remains cirrhotic even after a very protracted recovery period [55]. Our study indicates the same process after 9-month recovery period, liver fibrosis remain existing. In the other hand, it means a long term therapeutic window using this model.
CONCLUSION
Liver fibrosis represents a classical outcome of many chronic liver diseases. Animal models are being used for several decades to study fibrogenesis and to evaluate the anti-fibrotic potential of therapies and strategies. Previous study demonstrated that monkeys and human have similar liver architecture including hepatocyte, portal regions, bile duct, portal vein and liver veins [66]. Our study showed that liver fibrosis could be established by only given CCl4, which testify the hypothesis. In current stage, many technology could assist diagnose liver fibrosis, but no one indicator can diagnosis the diseases except for pathological result. The monkey model is a better system to explore the prevention and treatment of chronic liver diseases and develop new diagnostic techniques and novel treatment.
REFERENCES
- Anthony PP, Ishak KG, Nayak NC, Poulsen HE, et al. (1977). The morphology of cirrhosis: definition, nomenclature, and classification. Bull World Health Organ. 55(4): 521- 540.
- Friedman SL. (2008). Mechanisms of hepatic fibrogenesis. Gastroenterology. 134(6): 1655-1669.
- Hiromitsu Hayashi and Takao Sakai. (2011). Animal models for the study of liver fibrosis: new insights from knockout mouse models. Am J PhysiolGastrointest Liver Physiol. 300(5): G729-G738.
- Friedman SL. (2004). Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol. 1(2): 98-105.
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, et al. (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 3(5): 349-363.
- Tianhui Liu, Xiaoming Wang, Morten Karsdal, Diana Leeming, et al. (2012). Molecular Serum Markers of Liver Fibrosis. Biomarker Insights. 7: 105-117.
- Thomas A. Wynn. (2007). Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 117(3): 524-529.
- Wynn TA. (2008). Cellular and molecular mechanisms of fibrosis. J Pathol. 214(2): 199-210.
- Gines P, Cardenas A, Arroyo V and Rodes J. (2004). Management of cirrhosis and ascites. N. Engl. J. Med. 350(16): 1646-1654.
- Davis GL, Albright JE, Cook SF and Rosenberg DM. (2003). Projecting future complications of chronic hepatitis C in the United States. Liver Transpl. 9: 331-338/a>
- Iredale JP. (2003). Cirrhosis: new research provides a basis for rational and targeted treatments. BMJ. 327(7407): 143-147.
- Fallowfield JA and Iredale JP. (2004). Targeted treatments for cirrhosis. Expert Opin Ther Targets. 8(5): 423-435.
- Friedman SL, Maher JJ, Boyles JK, Yamasaki G, et al. (1992). Isolated hepatic lipocytes and Kupffer cells from normal human liver: morphological and functional characteristics in primary culture. Hepatology. 15(2): 234-243.
- Geerts A. (2001). History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 21(3): 311-335.
- Benyon RC, Iredale JP, Goddard S, Winwood PJ, et al. (1996). Increased expression of tissue inhibitor of metalloproteinases-1 and -2 relative to interstitial collagenase in fibrotic human liver. Gastroenterology. 110: 821-831.
- Constandinou C, Henderson N, and Iredale JP. (2005). Modelling liver fibrosis in rodents. Methods Mol. Med. 117: 237-250./a>
- Tsukamoto H, Matsuoka M and French SW. (1990). Experimental models of hepatic fibrosis: a review. Semin Liver Dis. 10(1): 56-65.
- Starkel P and Leclercq IA. (2011). Animal models for the study of hepatic fibrosis. Best Practice & Research Clinical Gastroenterology. 25(2): 319-333.
- Heinrich S, Georgiev P, Weber A, Vergopoulos A, et al. (2011). Partial bile duct ligation in mice: a novel model of acute cholestasis. Surgery. 149(3): 445-451
- Gershwin ME, Oertelt S, Lian ZX, Cheng CM, et al. (2006). Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-β receptor II dominant-negative mice. J Immunol. 177(3): 1655-1660.
- Gershwin ME, Wakabayashi K, Lian ZX, Moritoki Y, et al. (2006). IL-2 receptor a-/- mice and the development of primary biliary cirrhosis. Hepatology. 44(5): 1240-1249
- Sullivan BP, Cui W, Copple BL and Luyendyk JP. (2012). Early growth response factor-1 limits biliary fibrosis in a model of xenobiotic-induced cholestasis in mice. ToxicolSci. 126(1): 267-274.
- Trauner M, Fickert P, Stoger U, Fuchsbichler A, et al. (2007). A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am J Pathol. 171(2): 525-536.
- Slater TF, Cheeseman KH and Ingold KU. (1985). Carbon tetrachloride toxicity as a model for studying freeradical mediated liver injury. Philos Trans R SocLond B BiolSci. 311(1152): 633-645.
- Ding Z and Zhuo L. (2013). Attenuation of hepatic fibrosis by an imidazolium salt in thioacetamide-induced mouse model. J GastroenterolHepatol. 28(1): 188-201.
- Jenkins SA, Grandison A, Baxter JN, Day DW, et al. (1985). A dimethylnitrosamine-induced model of cirrhosis and portal hypertension in the rat. J Hepatol. 1(5): 489-499.
- Rinella ME and Green RM. (2004). The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. 40(1): 47-51.
- Sahai A, Malladi P, Pan X, Paul R, et al. (2004). Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol. 287(5): G1035-G1043.
- Tsukamoto H, Towner SJ, Ciofalo LM and French SW. (1986). Ethanol-induced liver fibrosis in rats fed high fat diet. Hepatology. 6(5): 814-822.
- Baba Y, Saeki K, Onodera T and Doi K. (2005). Serological and immunohistochemical studies on porcine-seruminduced hepatic fibrosis in rats. ExpMolPathol. 79(3): 229- 235.
- Yang SQ, Lin HZ, Lane MD, Clemens M and Diehl AM. (1997). Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl AcadSci U S A. 94(6): 2557-2562.
- Lin S, Lin Y, Nery JR, Urich MA, et al. (2014). Comparison of the transcriptional landscapes between human and mouse tissues. Proc Natl AcadSci USA. 111(48). 17224- 17229.
- Ziegler-Heitbrock L, Randolph GJ, Ingersoll MA, Spanbroek R, et al. (2010). Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. 115(3): e10-e19.
- Zheng-Jie Xu, Jian-Gao Fan, Xiao-Dong Ding, Liang Qiao, et al. (2010). Characterization of High-Fat, Diet-Induced, Non-alcoholic Steatohepatitis with Fibrosis in Rats. Dig Dis Sci. 55(4): 931-940.
- Weizhi Ji, Wei Si, Hong Wang, Junfeng Wang, et al. (2015). Rhesus monkey model of liver disease reflecting clinical disease progression and hepatic gene expression analysis. Scientific Reports. 5: 15019/a>
- K Ding, Liu MR, Huang K, Liang Y, et al. (2014). Establishment of a liver fibrosis model in cynomolgus monkeys. Experimental and Toxicologic Pathology. 66(5-6): 257-261.
- Bedossa P and Poynard T. (1996). The French METAVIR Cooperative Study Group. An algorithm for grading activity in chronic hepatitis C. MEDLINE Abstract. Hepatology. 24(2): 289-293.
- Liedtke C, Luedde T, Sauerbruch T, Scholten D, et al. (2013). Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis & Tissue Repair. 6(1): 19.
- Ahmad W, Ijaz B, Gull S, Asad S, et al. (2011). A brief review on molecular, genetic and imaging techniques for HCV fibrosis evaluation. Virol J. 8(1): 53.
- Zhou K and Lu LG. (2009). Assessment of fibrosis in chronic liver diseases. J Dig Dis. 10(1): 7-14.
- Daxboeck F, Gattringer R, Mustafa S, Bauer C, et al. (2005). Elevated serum alanine aminotransferase (ALT) levels in patients with serologically verified Mycoplasma pneumoniae pneumonia. ClinMicrobiol Infect. 11(6): 507-510.
- Giannini E, Risso D, Botta F, Chiarbonello B, et al. (2003). Validity and clinical utility of the aspartate aminotransferasealanine aminotransferase ratio in assessing disease severity and prognosis in patients with hepatitis C virusrelated chronic liver disease. Arch Intern Med. 163(2): 218-224.
- Lohr M, Hummel F, Martus P, Cidlinsky K, et al. (1999). Serum levels of extracellular matrix in acute pancreatitis. Hepatogastroenterology. 46(30): 3263-3270.
- Friedman SL. (2008). Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 88: 125-172.
- Tacke F and Weiskirchen R. (2012). Update on hepatic stellate cells: pathogenic role in liver fibrosis and novel isolation techniques. Expert Rev GastroenterolHepatol. 6(1): 67-80.
- Bataller R and Brenner DA. (2005). Liver fibrosis. J Clin Invest. 115(2): 209-218.
- Leeming DJ, Nielsen MJ, Dai Y, Veidal SS, et al. (2012). Enzyme-linked immunosorbent serum assay specific for the 7S domain of Collagen Type IV (P4NP 7S): A marker related to the extracellular matrix remodeling during liver fibrogenesis. Hepatol Res. 42(5): 482-493.
- Rosenberg WM, Voelker M, Thiel R, Becka M et al. (2004). Serum markers detect the presence of liver fibrosis: a cohort study. Gastroenterology. 127(6): 1704-1713.
- Stickel F, Poeschl G, Schuppan D, Conradt C, et al. (2003). Serum hyaluronate correlates with histological progression in alcoholic liver disease. Eur J GastroenterolHepatol. 15(9): 945-950.
- Fraser JR and Gibson PR. (2005). Mechanisms by which food intake elevates circulating levels of hyaluronan in humans. J Intern Med. 258(5): 460-466.
- Enrico Rossi, Leon A Adams, Max Bulsara and Gary P Jeffrey. (2007). Assessing Liver Fibrosis with Serum Marker Models. ClinBiochem Rev. 28(1): 3-10.
- Roeb E, Behrmann I, Grotzinger J, Breuer B, et al. (2000). An MMP-9 mutant without gelatinolytic activity as a novel TIMP-1 antagonist. FASEB J. 14(12): 1671-1673.
- Arthur MJ. (2002). Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology. 122(5): 1525-1528.
- Issa R, Zhou X, Constandinou CM, Fallowfield J, et al. (2004). Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross linking. Gastroenterology. 126(7): 1795- 1808.
- Abdel-Aziz G, Lebeau G, Rescan PY, B. Clement, et al. (1990). Reversibility of hepatic fibrosis in experimentally induced cholestasis in rat. Am J Pathol. 137: 1333-1342
- Afdhal NH and Nunes D. (2004). Evaluation of liver fibrosis: a concise review. Am. J. Gastroenterol. 99(6): 1160-1174
- Ratziu V, Charlotte F, Heurtier A, Gombert S, et al. (2005). Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology. 128(7):1898-1906.
- Thampanitchawong P and Piratvisuth T. (1999). Liver biopsy: complications and risk factors. World J. Gastroenterol. 5(4): 301-304.
- ZD Goodman. (2007). Grading and staging systems for inflammation and fibrosis in chronic liver diseases. Journal of Hepatology. 47(4): 598-607.
- Ming-HuwiHorng. (2007). An ultrasonic image evaluation system for assessing the severity of chronic liver disease. Computerized Medical Imaging and Graphics. 31(7): 485- 491
- Manuel Romero-Gomez, Gomez-Gonzalez E, Madrazo A, Vera-Valencia M, et al. (2008). Optical Analysis of Computed Tomography Images of the Liver Predicts Fibrosis Stage and Distribution in Chronic Hepatitis C. HEPATOLOGY. 47(3): 810-816
- Aguirre DA, BehlingCA, Alpert E, Hassanein TI, et al. (2006). Liver fibrosis: noninvasive diagnosis with double contrast material-enhanced MR imaging. Radiology. 239(2): 425-437.
- Cohen EI, Wilck EJ and Shapiro RS. (2006). Hepatic imaging in the 21st century. Semin Liver Dis. 26(4): 363-372.
- Hussain SM and Semelka RC. (2005). Hepatic imaging: comparison of modalities. Radiol Clin North Am. 43(5): 929-947.
- . Pang R, Liu J, He J, Wang H, et al. (2005). Establishment and evaluation of macaque liver fibrosis model. World Chin J Digestol. 13(16): 1956-1958.