Information Links
Related Conferences
Previous Issues Volume 10, Issue 1 - 2025
A Theory of Huntington's Disease: Phenocopy, Frame Shift and Rate Equation
Eugen Tarnow*
18-11 Radburn Road, Fair Lawn, NJ 07410, USA
*Corresponding Author: Eugen Tarnow, 18-11 Radburn Road, Fair Lawn, NJ 07410, USA, Phone: +16462290787, Email: [email protected]
Received Date: October 08, 2024
Published Date: January 03, 2025
Citation: Tarnow E. (2025). A Theory of Huntington's Disease: Phenocopy, Frame Shift and Rate Equation. Mathews J Psychiatry Ment Health. 10(1):48.
Copyrights: Tarnow E. (2025).
ABSTRACT
Huntington’s disease (HD) is one of the most well defined “repeat diseases”, associated with a short repeated genetic sequence, CAG. Recent research points to the possibility that HD is caused by a frame shift. Here is presented three pieces of evidence that supports the frame shift hypothesis. First, taking into account that a phenocopy of HD has a different repeat that is associated with a different gene, I suggest that the Huntingtin gene is not important for HD, only the repeat sequence is. Second, taking into account that a phenocopy of HD has a CTG repeat rather than a CAG repeat, and that the toxin should be the same for both disease types and that the third base in a codon is the least important, I propose the reading frame is shifted for the repeat expansions and the A/T difference occurs on the least important third base. The most likely sense and antisense reading frames are then (GCA)n and (GCT)n and the corresponding amino acid is polyalanine as previously suggested. Third, I suggest that the age of onset relationship follows a rate equation based on the frame shift. If the concentration is proportional to the probability of creating a polyalanine of length m in a repeat expansion of length n, the corresponding equation is borne out by the data on age of onset and repeat length and m is found to be about 30.6 (the onset for infinite age).
Keywords: Huntington’s Disease, CAG, Kinetics, Repeat Expansions.
INTRODUCTION
Because neurons are not regenerated, neurodegenerative diseases are different from other diseases in that an extremely slow and unlikely process can, after a lifetime, become deadly.
Huntington’s disease (HD) is one of more than 20 genetic repeat diseases [6]. The repeated sequence is CAG attached to the HTT gene. There is also a phenocopy (HDL2) with a repeat sequence CTG attached to the JPH3 gene as well as other genetic causes [7].
It is thought that the repeat sequence “results in production of HTT protein with an expanded polyglutamine tract (polyQ), leading to pathogenic HTT protein conformers that are resistant to protein turnover, culminating in cellular toxicity and neurodegeneration” [8], however this is controversial, see [9,10]. It seems unlikely that this explains the CTG phenocopy since they are located next to two different genes (HTT and JPH3) and CTG codes for leucine, not glutamine and glutamine is polar and leucine is non-polar.
A second problem with the theory of “pathogenic HTT protein conformers” is as follows. If a protein misfolds, it would seem reasonable that there should be a critical number of repeats that causes a misfold and more repeats beyond this critical number should have little effect. In HD, however, the age of onset is a monotonic function of the number of repeats and there is no evidence of a critical number of repeats other than the age of onset increases: extrapolation suggests that at 37 repeats the median age of onset should be 79 and at 35 it should be 95 (Figure 1, data from Brinkman et al. [4]).
.png)
Figure 1. HD median age of onset (50% of individuals affected) as a function of the repeat length. Data from Brinkman et al. [4], fitted by the current author. The line on the left is an extrapolation of the data.
In this paper I will propose three new ideas that support the frame shift hypothesis [1,2] for the HD toxin. Together they identify the toxin to be polyalanine (present in the Huntington brain, in particular where there is neuronal loss, according to [2] before repeated about 30.6 times.
SAMPLE & METHOD
The data used is from Brinkman et al. [4] who describes it:
“For the purpose of this study we used those individuals with CAG expansions of the upper allele that were >28, comprising 728 affected and 321 asymptomatic at-risk individuals from 473 families, whose age at onset or oldest age while still asymptomatic could be ascertained.”
“An accurate assessment of the age at onset was performed through both a retrospective review of patient charts and telephone interviews with patients, family members, genetic counselors, and physicians. Age at onset was defined as the first time at which a patient had either neurological or psychiatric symptoms that represented a permanent change from the normal state. The age used for analysis of all asymptomatic individuals was the oldest age when his or her clinical status was last directly confirmed, either at the genetics clinic in Vancouver or by the local, attending physician. Particular attention was paid to confirmation of current age and clinical status of all asymptomatic, at-risk individuals in the HD database who were >65 years of age.”
Microsoft Office Excel 2007 was used to curve fit the median age as a function of the number of CAG repeats.
Results: It is the CAG expansion, not the gene
If we assume that HD and its phenotype copy has the same pathology, it means that it is the expansion, not the different neighboring genes HTT and JPH3, that is responsible for the pathology. This is particularly true if we insist that the two expansions are the same as I do in the next section.
Results: Rule for Reading Frame Assignment
I propose that for repeat diseases with phenocopies that have shifted repeats, reading frame errors are crucial and that the relevant reading frames are those that cause both repeats to code for the most alike protein. For a three-base codon repeat sequence in which the phenocopy has a single base substituted, the relevant reading frame is the one in which the substitution occurs on the third codon base, the least important one.
Thus for the CAG and CTG expansions of HD and its phenocopy, the reading frame is changed so that the codon base substitution occurs in the third base instead of the second: instead of (CAG)n and (CTG)n it is (GCA)n and (GCT)n. Both GCA and GCT code for the same amino acid – alanine.
I next suggest that the correlation of repeats with HD age of onset may be the kinetic equation of the corresponding biochemical reaction creating the toxin. Such a slow reaction arising from an unlikely reading frame error may be at the heart of HD.
Results: A Clue from Reaction Kinetics
I suggest that the relationship between repeat length and age of onset can be reframed in terms of chemical kinetics and assume that the onset of the disease happens at a fixed critical concentration:
rate of reaction*age=critical concentration of toxin (equation 1)
Next I assume that the rate limiting step is the reading frame error resulting in a poly alanine molecule of length m. There are n-1-m positions in which this reading frame error can occur (because of the shift there are at most n-1 positions and in order to fit m alanines the number of positions is limited to n-1-m), the probability of creating this molecule is proportional to n-1 -m:
(n-1-m)*age=const
or
n=1+m+const/age (equation 2)
or
1/age=(n-1-m)/const
If I replot the data points of Figure 1 in Figure 2 with the y axis as 1/age the curve fulfills equation 2 and I identify m=30.6:
.png)
Figure 2. The inverse of the (age of onset) versus the length of the repeat. The intersection with the x-axis identifies the length of the poly-alanine to be about 30.6 alanine units.
I calculate the age of onset as a function of n and find the following formula:
Age of onset = 505/(n-31.6) (equation 3)
505 corresponds to the critical concentration of polyalanine divided by the rate of the reading frame error with the n and m dependency removed. This is an alternative to the exponential fit proposed by Kaplan et al. [11] but, in the mind of this author, not borne out by their theory. The two fits are compared in Figure 3.
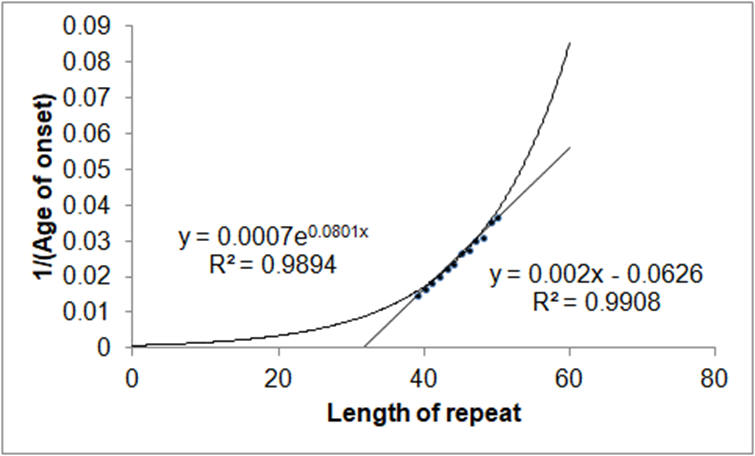
Figure 3. Exponential fit and the fit from equation 3 compared. To distinguish the two fits, and perhaps settle the kinetics, it may be necessary to probe the extremes of repeats at less than 39 and more than 50.
DISCUSSION
Supporting other authors that the HTT gene itself is not important I noted there is a phenocopy of HD in which the repeat expansion occurs at a different gene (JPH3 instead of HTT). Also since the repeat expansions of HD and its phenocopy are somewhat different, this would limit the toxin to frameshifted polyalanines. This mechanism has been earlier proposed to account for Machado–Joseph disease [1] and also to account for Huntington’s disease (previously proposed by, after for example [2]). I interpreted, for the first time, the relationship between the repeat length and the age of onset as a kinetic equation for the creation of the polyalanine. I converted the repeat length into a concentration by noting that the probability of creating a frameshifted polyalanine of length m in a repeat expansion of length n is proportional to n-1-m. I showed that the corresponding linear equation between the inverse of the age of onset and the length of the repeat n is borne out by the data and allows me to identify the repeat length of the HD toxic polyalanine to be 30.6.
There is previous literature that also suggests that the Huntingtin gene is not important: Lee et al. [3] arrived at this conclusion in a completely different way and write that “The effects of variable, glutamine-encoding, CAA interruptions indicate that a property of the uninterrupted HTT CAG repeat sequence, distinct from huntingtin’s polyglutamine segment, dictates the rate at which HD develops”. Differently, “The number of consecutive glutamines in huntingtin is not equal to the number of consecutive CAG repeats because CAA interruptions also code for glutamine. Consequently, two individuals with the same CAG repeat length can produce different numbers of consecutive glutamines in their huntingtin protein, depending on the presence or absenceof CAA codons. The length of the uninterrupted CAG repeat turns out to be a better predictor of age-at-onset than is the length of the polyglutamine tract, indicating that the driver of the rate at which HD develops is a property of the uninterrupted CAG repeat rather than the length of encoded polyglutamine.” (James Gusella, one of the authors of the Lee et al, 2019, paper, private communication). This is also consistent with my point that the toxin is not polyglutamine but polyalanine. Note that the frameshifted CAA would be ACA which codes for threonine, not alanine, and would consequently break up the polyalanine.
There is previous literature showing that HD includes frameshifted products including polyalanine [2,12] and the toxicity of polyalanines has been reviewed elsewhere [12].
A polyalanine molecule of about 30.6 units provides the first explanation as to why at least 36 repeats are needed for HD to penetrate: several fewer repeats than 36 would make the creation of a 30 or 31 unit polyalanine molecule impossible with the reaction proposed and since the reaction creating the toxin is proportional to (n-1-m), the closer we get to m=n the much slower the reaction proceeds.
Graveland et al. [14] write that “the morphology of dendrites of medium-sized spiny neurons was markedly altered by the appearance of recurved endings and appendages, a decrease or increase in the density of spines, and abnormalities in the size and shape of spines”. Is it possible that this is caused by a polyalanine repeat?
In at least one other neurodegenerative disease the most glaring pathogen is not the toxin: In Alzheimer’s disease, amyloid plaques, the most obvious pathology thought to be toxic to neurons and thought to appear in late life coinciding with the dementia, were removed from patients but did not result in any clinical improvement [15,16]. A new suggestion is that Alzheimer’s disease begins already in childhood or in puberty not with amyloid plaques but with abnormal tau protein deposits in an extremely slow process [17]. It may be that HD is similarly not caused by the HTT protein with an expanded polyglutamine tract but rather by a previously unlikely candidate resulting from an extremely slow and unlikely reading frame shift.
If the HD toxin is indeed caused by frame shifting, it is the first identified disease in which frame shifting is the underlying cause. Other diseases in which frame shifting is a factor including some cancers, Chron’s disease, cystic fibrosis, HIV, etc [18-23].
Problems with this theory include:
- It does not explain why homozygotes have the same age of onset as heterozygotes, rather my theory suggests that homozygotes should have half the age of onset for the same repeat length N Perhaps the polyalanine becomes chiral and the chirality matters.
- It does not explain why other CAG repeat diseases have different symptoms Perhaps the protein gene next to the repeat sequence matters in these diseases.
REFERENCES
- Gaspar C, Jannatipour M, Dion P, Laganière J, Sequeiros J, Brais B, et al. (2000). CAG tract of MJD-1 may be prone to frameshifts causing polyalanine accumulation. Hum Mol Genet. 9(13):1957-1966.
- Bañez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, et al. (2015). RAN Translation in Huntington Disease. Neuron. 88(4):667-677.
- Lee JM, Correia K, Loup J, Kim KH, Barker D, Hong EP, et al. (2019). Huntington's disease onset is determined by length of uninterrupted CAG, not encoded polyglutamine, and is modified by DNA maintenance mechanisms. bioRxiv:529768.
- Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. (1997). The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet. 60(5):1202.
- Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, et al. (2004). Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A. 101(10):3498-3503.
- La Spada AR, Taylor JP. (2010). Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 11(4):247-258.
- Mariani LL, Tesson C, Charles P, Cazeneuve C, Hahn V, Youssov K, et al. (2016). Expanding the Spectrum of Genes Involved in Huntington Disease Using a Combined Clinical and Genetic Approach. JAMA Neurol. 73(9):1105-1114.
- Dickey AS, La Spada AR. (2018). Therapy development in Huntington disease: From current strategies to emerging opportunities. Am J Med Genet A. 176(4):842-861.
- Ratovitski T, Chaerkady R, Kammers K, Stewart JC, Zavala A, Pletnikova O, et al. (2016). Quantitative Proteomic Analysis Reveals Similarities between Huntington's Disease (HD) and Huntington's Disease-Like 2 (HDL2) Human Brains. J Proteome Res. 15(9):3266-3283.
- Veitch NJ, Ennis M, McAbney JP; US-Venezuela Collaborative Research Project; Shelbourne PF, Monckton DG. (2007). Inherited CAG.CTG allele length is a major modifier of somatic mutation length variability in Huntington disease. DNA Repair (Amst). 6(6):789-796.
- Kaplan S, Itzkovitz S, Shapiro E. (2007). A universal mechanism ties genotype to phenotype in trinucleotide diseases. PLoS Comput Biol. 3(11):e235.
- Toulouse A, Au-Yeung F, Gaspar C, Roussel J, Dion P, Rouleau GA. (2005). Ribosomal frameshifting on MJD-1 transcripts with long CAG tracts. Hum Mol Genet. 14(18):2649-2660.
- Amiel J, Trochet D, Clément-Ziza M, Munnich A, Lyonnet S. (2004). Polyalanine expansions in human. Hum Mol Genet. 13(2):R235-R243.
- Graveland GA, Williams RS, DiFiglia M. (1985). Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 227(4688):770-773.
- Drachman DA. (2014). The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer's disease. Alzheimers Dement. 10(3):372-380.
- Giacobini E, Gold G. (2013). Alzheimer disease therapy--moving from amyloid-β to tau. Nat Rev Neurol. 9(12):677-686.
- Braak H, Del Tredici K. (2012). Where, when, and in what form does sporadic Alzheimer's disease begin? Curr Opin Neurol. 25(6):708-714.
- Deckel AW, Volmer P, Weiner R, Gary KA, Covault J, Sasso D, et al. (2000). Dietary arginine alters time of symptom onset in Huntington's disease transgenic mice. Brain Res. 875(1-2):187-195.
- Leopold NA, Podolsky S. (1975). Exaggerated growth hormone response to arginine infusion in Huntington's disease. J Clin Endocrinol Metab. 41(1):160-163.
- Podolsky S, Leopold NA. (1977). Abnormal glucose tolerance and arginine tolerance tests in Huntington's disease. Gerontology. 23(1):55-63.
- Saudou F, Humbert S. (2016). The Biology of Huntingtin. Neuron. 89(5):910-926.
- Winter R, Liebold J, Schwarz E. (2013). The unresolved puzzle why alanine extensions cause disease. Biol Chem. 394(8):951-963.
- Wojciechowska M, Olejniczak M, Galka-Marciniak P, Jazurek M, Krzyzosiak WJ. (2014). RAN translation and frameshifting as translational challenges at simple repeats of human neurodegenerative disorders. Nucleic Acids Res. 42(19):11849-11864.