Information Links
Related Conferences
Previous Issues Volume 8, Issue 1 - 2023
A Rare Case of Childhood Adrenocortical Carcinoma in a 4-Years-Old Female Child Presented with Features of Cushing Syndrome: Case Report and Literature Review
Gashaw Arega1*, Daniel Hailu1, Abdulkadir Mohammedsaid Gidey1, Abel Hailu1, Mulualeme Nigusie1, Getasew Fikad1, Sewagegnh Yeshiwas2, Abrehet Zeray3, Samuel Sisay3
1Division of Hematology and Oncology, Department of Pediatrics and Child Health, Addis Ababa University, Addis Ababa, Ethiopia
2Pediatric Endocrinology Unit, Department of Pediatrics and Child health, Addis Ababa University, Addis Ababa, Ethiopia
3Pediatrics Radiology Unit, Department of Radiology, Addis Ababa University, Addis Ababa, Ethiopia
*Correspondence: Gashaw Arega, Department of pediatrics and child health, School of Medicine, Addis Ababa University, Addis Ababa, Ethiopia; Tel: +251911417235 Email: [email protected]
Received Date: January 13, 2023
Publication Date: January 27, 2023
Citation: Arega G, et al. (2023). A Rare Case of Childhood Adrenocortical Carcinoma in a 4-Years-Old Female Child Presented with Features of Cushing Syndrome: Case Report and Literature Review. Mathews J Cancer Sci. 8(1):36.
Copyright: Arega G, et al. © (2023).
ABSTRACT
Childhood adrenocortical carcinoma is an extremely rare cancer with a poor prognosis. It usually presents during the first 5 years of life with a median age of 3–4 years, although there is a second smaller peak during the adolescence period. Here, we report a 4 – years-old female child diagnosed with childhood adrenocortical carcinoma with distant metastasis after she presented with features of Cushing syndrome and recent worsening of abdominal swelling of month duration. Chest and abdomen CT scan showed a left adrenal mass with liver and lung metastasis. Serum ACTH was low and serum cortisol was high. Biopsy from the liver showed secondary deposits with malignant carcinoma. She started treatment with chemotherapy with EDP regimen with palliative intent. Despite chemotherapy she had progression of disease with systemic multiorgan involvement.
Keywords: Childhood adrenocortical carcinoma, Cushing syndrome, Distant metastasis, Edp regimen
INTRODUCTION
Childhood adrenocortical carcinoma is a rare cancer with a poor prognosis [1,2]. The incidence of adrenocortical tumors in children is extremely low, accounting only 0.2% of pediatric cancers [3]. Childhood adrenocortical tumors typically present during the first 5 years of life with a median age of 3–4 years, although there is a second, smaller peak during adolescence [4-6]. In the pediatric population, there is a predilection for the female gender the ratio of female to male ranges from 1.5–2.5:1 [2,7]. Adrenocortical tumors are typically large tumors upon clinical presentation, often measuring more than 6 cm in diameter. The hallmark of a biochemical evaluation of an adrenocortical tumor is the measurement of steroid hormones produced by the tumor. Most patients with cortisol-secreting tumors will have suppressed ACTH (10 pg/mL) and increased cortisol on a spontaneous 8:00 AM blood draw [8]. Our patient clinically presented with excessive hair growth over the pubic area, axilla, and trunk. She had Cushing features with significant weight gain and hyperpigmentation with papular non-itchy rash over the skin. On evaluation, she had moon appearance face, acne, hirsutism, adult pattern pubic hair growth, stage I hypertension, and a large predominantly left flank mass extending up to the left upper quadrant and mid abdomen. On investigation she had low serum ACTH and hypercortisolism, imaging with CT showed a left adrenal heterogenous mass with surrounding organ invasion and distant lung and liver metastasis. Biopsy from the hepatic deposits showed malignant carcinoma with necrosis and mitosis. A diagnosis of Stage IV adrenocortical carcinoma was made and the child started on combination chemotherapy with EDP regimen with palliative intent.
CASE PRESENTATION
A 4 year-old -female child presented with excessive hair growth initially over the pubic area and later involving the face, trunk, and axilla of a year duration. Four-months after these complaints, she began to developa a pinkish, non-itchy skin rash over the face and back. She also had significant weight gain. And later the family noticed an abdominal swelling with gradual increment and showed singnigicant increment over a month. And they sought medical care at a tertiary hospital when the abdominal swelling increases in size.
On physical examination, she had a moon face appearance and vital signs revealed a pulse rate of 110 beats per minute, respiratory rate of 22 breaths per minute, a febrile, maintained saturation of oxygen in atmosphere air and blood pressure of 110/70 mm Hg, which was stage I hypertension for her age. On anthropometry, her weight was 25 kg, height-110cms, and mid-upper arm circumferences (MUAC) were 21cms. Weight for height was >3Z score and she had childhood obesity. She had no enlarged lymph nodes in all accessible areas, no thyroid and breast enlargement (pre-adolescent, Tanner stage 1). On other pertinent physical examination, she had a distended abdomen with everted umbilicus; the liver was palpated 10 cm below the right coastal margin with a total liver span of 16 cm with surface. There was a 14 by 10 cm non-tender, firm palpable mass over the left upper quadrant of the abdomen predominately over the flank area.
On the genitourinary system, the child had adult pattern pubic hair (Tanner stage 5) and clitoromegaly (Figure 1). On the integumentary system, she had acne over the face, multiple hyper-pigmented papular lesions, and hirsutism all over her face and back.

Figure 1:Adult pattern pubic hair in a 4-years-old female child diagnosed with childhood adrenocortical carcinoma.
Laboratory investigations revealed a normal complete blood count (CBC) with differential and normal renal function test with creatine of 0.3 mg/dl. Liver function tests showed ALT-39iu/l, AST-101 iu/L, and ALP-115iu/l. Serum electrolytes showed mild hypokalemia of 3.13 mmol/l. Thyroid function tests, serum FISH, and LH were in the normal range. Serum ACTH is 1.4 pg.ml [Reference range 7.2-63.3 ng/ml] and serum cortisol is high [Reference range 2.3-11.9ug/dl]. Bone marrow biopsy showed trilineage hematopoiesis.
Abdominal ultrasound showed a heterogenous left supra renal mass with multiple liver lesions. Chest and abdominal CT scan showed a 14.2cmx6.4cm well-defined heterogeneously enhanced left suprarenal mass with internal areas of chunky calcification and necrosis. The mass extended down anterior to the left kidney abutting the renal pedicle with no invasion or thrombus. It had pushed the left kidney inferiorly and posteriorly. The liver showed multiple large heterogeneously enhancing lesions with internal necrotic areas. There were also multiple peripheral nodules in the bilateral lung bases (Figure 2).
A. 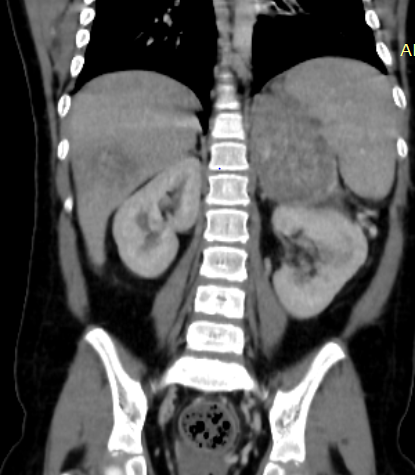 B.
B.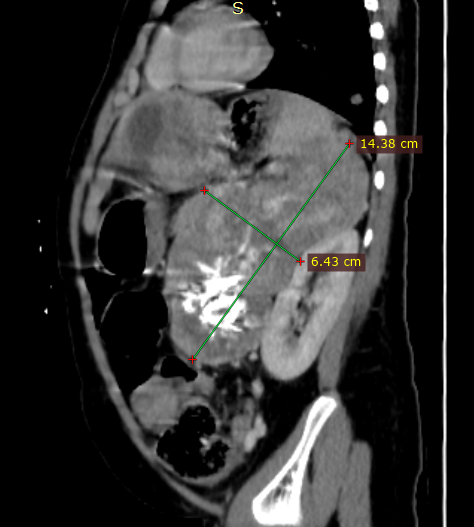
C. 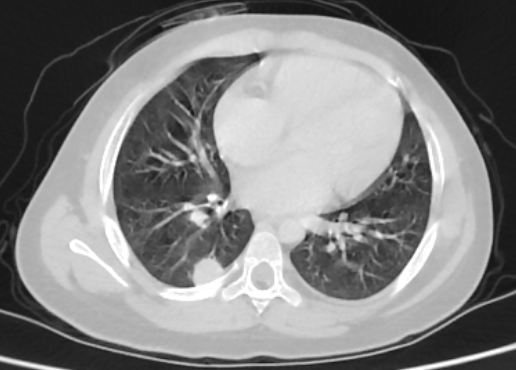 D.
D.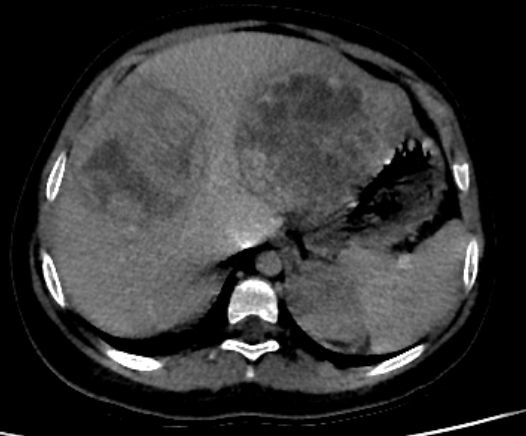
Figure 2: Coronal and sagittal reformatted contrast-enhanced CT image (A and B) shows large heterogeneously enhanced left suprarenal mass with calcification (black arrow). Note the liver metastasis in (white arrow in A). Axial CT image of the lung (C) demonstrates a peripherally located metastatic lesion in the right lung (white arrow). Axial contrast-enhanced CT image (D) shows a heterogenous left suprarenal mass (asterisk) with liver metastasis (white arrow).
Core needle biopsy was taken from the accessible hepatic mass and histopathology showed moderately pleomorphic polygonal to round cells having abundant eosinophilic to vacuolated cytoplasm and centrally located pleomorphic round to oval nuclei with prominent nucleoli arranged in a sheet and cord with intervening delicate fibrovascular stroma with focal area of necrosis and mitosis, which are suggestive of malignant carcinoma (Figures 3 and 4).
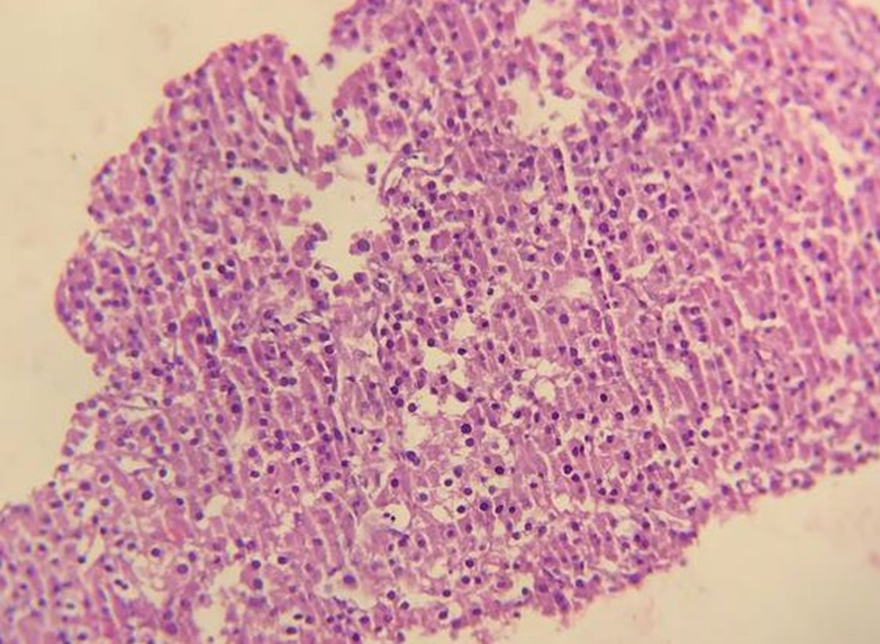
Figure 3: Histopathology shows polygonal to round cells having abundant eosinophilic to vacuolated cytoplasm and pleomorphic nuclei with prominent nucleoli arranged in sheets and trabeculae, 20X Objective.
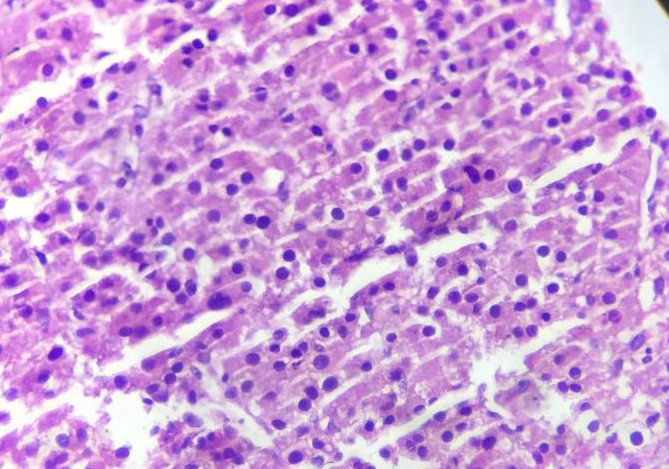
Figure 4: Histopathology shows polygonal to round cells having abundant eosinophilic to vacuolated cytoplasm and pleomorphic nuclei with prominent nucleoli arranged in sheets and trabeculae with mitotic activity, 40X Objective.
With the clinical presentation, low serum ACTH level and hypercortisolism, CT scan imaging, and core needle tissue biopsy core result, the diagnosis of childhood adrenocortical carcinoma with distant metastasis [Stage IV Child hood adrenocortical carcinoma] was made. Treatment options were discussed among the families and she was starting chemotherapy with EDP-M regimen with Etoposide, Doxorubicin, and Cisplatin. And since mitotane was not available, we used ketoconazole in place of it.
CASE DISCUSSION
Adrenocortical carcinoma is a rare cancer with a worse prognosis [1.2]. The incidence of adrenocortical tumors in children is extremely low, accounting only 0.2% of pediatric cancers [3]. Childhood adrenocortical tumors typically present during the first 5 years of life with a median age of 3–4 years, although there is a second, smaller peak during adolescence [4-6]. In the pediatric population, there is a predilection for the female gender (the ratio of female to male ranges from 1.5–2.5:1 [2,7].
Most patients with cortisol-secreting tumors have suppressed ACTH (10 pg/mL) and increased cortisol on a spontaneous 8:00 AM blood draw. The diagnosis of hypercortisolism is usually established by a 1 mg dexamethasone suppression test, midnight salivary cortisol, or elevated 24-hour urine-free cortisol [8]. Radiologically, adrenocortical tumors usually measure more than 6cms and vary in appearance with heterogenous enhancement due to the presence of necrosis, calcifications, and internal hemorrhage [9-11].
Invasion of adjacent organs and venous extension may present at the initial presentation. Metastasis to distant organs are common in advanced stage, usually to the liver, lungs, and lymph nodes [10]. Metastatic deposits, which are largely similar to primary adrenocortical tumors, both in terms of cellular histology and the absence of desmoplasia. Adrenocortical carcinoma deposits in the liver are often intimately intertwined with hepatocytes [12].
The stage of the disease, specifically the presence of distant metastasis and the number of organs involved in metastatic disease, confers a poor prognosis [13,14]. The treatment options depend on the stage of the disease and all therapies of metastatic adrenocortical carcinoma must be considered palliative [12]. For advanced adrenocortical carcinoma, the first line of chemotherapy therapy is EDP plus mitotane with better rate of response and progression-free survival [15].
Our patient clinically presented with excessive hair growth over the pubic area, axilla, and trunk. She also had Cushing features with significant weight gain and hyperpigmentation with papular non-itchy rash over the skin. On physical examination, she had a moon appearance face, acne, hirsutism, adult pattern pubic hair growth, stage I hypertension, and a large predominantly left flank mass extending up to the left upper quadrant and mid abdomen. On investigation, she had low serum ACTH and hypercortisolism, and mild hypokalemia.
Imaging with CT showed a large left adrenal heterogenous mass with surrounding organ invasion and distant lung and liver metastasis. The biopsy from the hepatic deposit showed pleomorphic polygonal to round cells with centrally located pleomorphic round to oval nuclei with prominent nucleoli arranged in a sheet and cords with focal areas of necrosis and mitosis suggestive of malignant carcinoma. In our patient with the clinical presentation, low serum ACTH level and hypercortisolism, CT scan imaging, and core needle tissue biopsy result, the diagnosis of childhood adrenocortical carcinoma with distant metastasis [Stage IV Child hood Adrenocortical carcinoma] was made. And with the discussion with the family, chemotherapy with EDP-M regimen (Etoposide, Doxorubicin and Cisplatin and Mitotane) started with Palliative intent. Despite two cycles of chemotherapy, she had clinical worsening with new onset of seizure, deranged liver function tests, and worsening of chest conditions. And she passed away due to the disease progression with multiorgan involvement.
CONCLUSION
Adrenocortical carcinoma is a very rare cancer in childhood. Initial evaluation should be guided by the presenting history and physical examination findings; hirsutism, Cushing syndrome, and elevated blood pressure with or without hypokalemia. Biochemical evaluation of steroid hormones, imaging with CT scan/MRI, and biopsy of the mass helps to establish the confirmatory diagnosis of adrenocortical carcinoma. The treatment depends on the stage of the disease, for the advanced disease, a combination of chemotherapy with etoposide, doxorubicin, cisplatin, and mitotane is the first line of therapy. Since adrenocortical carcinoma is an extremely rare cancer with poor prognosis, a high index of suspicion is important to detect at the early stage of the disease.
DECLARATIONS
Ethical Clearance
Written informed consent for publication of their details, including the pictures and radiological images, was obtained from the parents. In addition, ethical clearance was obtained from the institutional review of Tikur Anbessa Specialized Hospital and Addis Ababa University.
Competing Interests
There are no conflicts of interest.
Authors' Contributions
Gashaw Arega, Daniel Hailu, Abdulkadir Mohammedsaid, Abel Hailu, Mulualeme Nigusie, and Getasew Fikad wrote the detailed oncology summary of the case. Sewagegnh Yeshiwas wrote the endocrinology part of the case. Abrehet Zeray, and Samuel Sisay wrote the radiology part. All the authors reviewed the manuscript and approved for the publication.
Funding
Nil.
Availability of Data and Materials
The data underlying this article are available in the article and in its online supplementary material.
REFERENCES
- Wooten MD, King DK. (1993). Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer. 72(11):3145-3155.
- Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG, et al. (2004). Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 22(5):838-845.
- Wieneke JA, Thompson LD, Heffess CS. (2003). Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol. 27(7):867-881.
- Redlich A, Boxberger N, Strugala D, Frühwald MC, Leuschner I, Kropf S, et al. (2012). Systemic treatment of adrenocortical carcinoma in children: data from the German GPOH-MET 97 trial. Klin Padiatr. 224(6):366-371.
- Gulack BC, Rialon KL, Englum BR, Kim J, Talbot LJ, Adibe OO, et al. (2016). Factors associated with survival in pediatric adrenocortical carcinoma: An analysis of the National Cancer Data Base (NCDB). J Pediatr Surg. 51(1):172-177.
- Berstein L, Gurney JG. (1999). Carcinomas and other malignant epithelial neoplasms. In: Ries LA, Smith MA, Gurney JG, et al. (eds). Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program. 11:139-148.
- Luton JP, Cerdas S, Billaud L, Thomas G, Guilhaume B, Bertagna X, et al. (1990). Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med. 322:1195-1120.
- Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. (2008). The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 93:1526-1540.
- Johnson PT, Horton KM, Fishman EK. (2009). Adrenal mass imaging with multidetector CT: pathologic conditions, pearls, and pitfalls. Radiographics. 29:1333-1351.
- Bharwani N, Rockall AG, Sahdev A, Gueorguiev M, Drake W, Grossman AB, et al. (2011). Adrenocortical carcinoma: the range of appearances on CT and MRI. AJRAm J Roentgenol. 196:W706-W714.
- Dunnick NR, Heaston D, Halvorsen R, Moore AV, Korobkin M. (1982). CT appearance of adrenal cortical carcinoma.J Comput Assist Tomogr. 6:978-982
- Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, et al. (2013). Adrenocortical Carcinoma. Endocr Rev. 35(2):293-294
- Drelon C, Berthon A, Ragazzon B, Tissier F, Bandiera R, Sahut-Barnola I, et al. (2012). Analysis of the role of Igf2 in adrenal tumour development in transgenic mouse models. PLoS One. 7:e44171.
- Hanahan D, Weinberg RA. (2000). The hallmarks of cancer. Cell. 100:57-70.
- Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, et al. (2012). Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 366(23):2189-2197.