Information Links
Related Conferences
Current Issue Volume 10, Issue 1 - 2025
Stargardt Disease a Clinical Finding of Circumb Papillary Band Shape Hyperpigmentery Hallow
Yaghoobi Gholamhossein*
Department of Ophthalmology, Razi Hospital, Ghafari St, Birjand University of Medical Sciences, Birjand, Iran
*Corresponding Author: Dr. Gholamhossein Yaghoobi, Department of Ophthalmology, Razi Hospital, Ghafari St, Birjand University of Medical Sciences, Birjand, Iran, Phone: 985632626251, Email: [email protected]
Received Date: October 10, 2025
Published Date: October 28, 2025
Citation: Gholamhossein Y. (2025). Stargardt Disease a Clinical Finding of Circumb Papillary Band Shape Hyperpigmentery Hallow. Mathews J Ophthalmol. 10(1):38.
Copyrights: Gholamhossein Y. © (2025).
ABSTRACT
Introduction: The Stargardt disease is the most common cause of decreased of vision in adults of less than 50 years old. This disease is mutations in the ABCA4 gene which is located in the chromosome chain 1 short sleeve. It works by encode the ATP-binding cassette (ABC) protein transporter expressed by the outer trunk cell segment. Case Presentation: A 12 years old girl brought to eye clinic by parent due to visual complain, visual acuity with correction of the right eye and left eye was 20/200 and fundoscopic finding showed the macular lesion indeed of flecks retina finding. The circumb papillary band hyperpigmentery hallow finding may be a characteristic sub type feature of stargardet disease. Conclusion: A case of Stargardt disease has been reported in a 12-year-old girl. The circumb papillary band hyperpigmentery hallow may be a characteristic sub type feature of stargardet disease. Althought it needed to report othere cases and confrmatory paraclinical documentation.
Keywords: Stargardt’s Disease, Case Report, Circumb Papillary Band Hyperpigmentery Hallow.
INTRODUCTION
Stargardet disease is the most common type of hereditary macular dystrophy. Although the incidence is not well known, estimates range from 1 in 8,000 to 10,000. The mode of inheritance is usually autosomal recessive, and mutations in the ABCA4 gene have been associated with the disease [1]. There is no gender or racial predilection, and it is heterogeneous in both clinical and genetic features. Patients usually present in the first or second decade of life with complaints of bilateral visual loss. Late onset is associated with a better prognosis. Positive central scotoma and changes in color perception may also be present. Visual acuity gradually declines to 6/60. Fundus examination reveals some yellow spots and the appearance of cochlear mucus in the macula in the early stages. The disease gradually progresses to bull's-eye maculopathy with retinal pigment epithelial atrophy or a tanned appearance in the late stages. The changes occur predominantly in the posterior pole, but may occasionally affect the peripheral retina, showing disc pallor, vascular attenuation, and pigmentary abnormalities [2,3].
Stargardt disease (SD) is one of the most common inherited macular dystrophy in both adults and children. This disease is a bilateral and symmetrical Maculopathy with autosomal recessive transmission linked to the ABCA4 gene, located on chromosome 1 in the region 1p22. It is both clinically and genetically highly heterogeneous. Age of onset is a surrogate marker: The earlier the onset, the more severe the disease course, with better prognosis generally associated with a later onset [4]. This case has a band of hyperpigmenty hollow around optic discs bilateraly.
CASE REPORT
A 12 years old girl who have visual complain attended by parent to eye clinic. The visual acuity with correction of the right eye and left eye was 20/200 and fundoscopic finding showed the macular lesion indeed of flecks retina finding. The circumb papillary band hyperpigmentery hallow finding also noted as showed in figure 1 so, this finding may be a characteristic sub type feature of stargardet disease. Although it needed more report. The characteristic Beaton bronze macular appearance in this case confirmed diagnosis of stargardet disease (Figure 1).
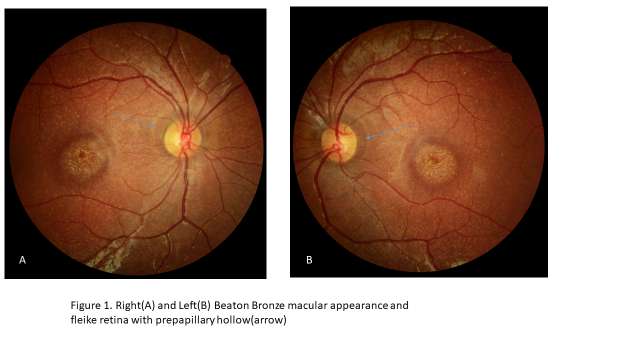
DISCUSSION
This case showed typically prepapillary hollow of hyperpigmentary change that it could be an early diagnostic sign although there is other report that indicate to save or no change of prepapillar chang of retinal pigment epithelium [5].
St diseases is the most common macular degeneration in children below 15 years old. This genetic disorder leads to a progressive and irreversible central vision loss, typically commencing between the ages of 7 and 15. While it diagnosed in youth, Stargardt's disease may have a similar feature such age-related maculopathy, causing diagnostic challenges in adults [4].
Therefore, if this clinical finding may be related to subtype it will be confirmatory by other finding in retinal finding or paraclinical such as OCT or gen detection.
Stargardt disease is one of the most common causes of inherited childhood and adulthood visual impairment. Stargardt disease is both phenotypically and genetically highly heterogeneous with significant advances having been made in our ability to identify the disease at the earliest stages, characterise clinical features that allow better-informed advice on prognosis, perform accurate rapid molecular genetic testing, and in our understanding of underlying disease mechanisms [6].
In the early stages of Stargardt disease, ophthalmoscopy may reveal a normal-appearing fundus. Due to the heterogeneity of Stargardt disease, providing a universal description of physical findings is difficult and often relies on imaging and ancillary studies.
Misdiagnosed STGD1 can sometimes lead to significant frustration in children due to lagging behind peers in school or kindergarten, declining reading levels, and the development of unsettling psychological, behavioral, and motor issues. These challenges are often compounded by diagnoses such as attention-deficit/hyperactivity disorder (ADHD) or dyslexia [7]. The early diagnosis as mention above could be preventative and guide to child and parent.
ACKNOWLEDGMENTS
None.
CONFLICT OF INTEREST
The author declares there are no conflicts of interest.
REFERENCES
- Delijani K, Sadowsky D, Baniadam K, Popovsky D, Sutariya R, Davis W. (2022). Stargardt Disease. Georgetown Medical Review. 6(1). DOI:10.52504/001c.36966.
- Bhakhri R, FAAO, Neidermann S, Vivacqua G. (2023). PEER REVIEWED Stargardt Disease: a Teaching Case Series. Optometric Education. 49(1).
- Harshika, Atul Mishra. (2023). Stargardt Disease: A Rare Case Report. International Journal Dental and Medical Sciences Research. 5(5):346-349.
- Hamza Lazaar M, Saadbenchekroun H, Facherkaoui AL. (2024). A Case Report: Exploring Stargardt Disease - Clinical Features and Genetic Insights. Int J Adv Res. 12(02):662-667.
- Bhakhri R, Neidermann S, Vivacqua G. (2023). Stargardt Disease: a Teaching Case Series. Optometric Education. 49(1).
- Maurya KS, Saxena S, Kumar V, Sinha M, Johny M, Patel R, et al. (2022). Stargardt Disease: A Case Study. WJPMR. 8(6):203-206.
- Ambrosio L, Perepelkina T, Elhusseiny AM, Fulton AB, Gonzalez Monroy JE. (2025). Advancing Insights into Pediatric Macular Diseases: A Comprehensive Review. J Clin Med. 14(2):614.