Information Links
Related Conferences
Previous Issues Volume 7, Issue 2 - 2023
Role of Biomarkers in Pathogenesis and Updated Treatment of Motor Neuron Disease: A Review Article
Ravi Kumar*, Arvind Kumar, Vimal Kumar Bharti, Bhuwanendra Singh, Kuldeep Saini, Sunita Jha, Minata
SD College of Pharmacy & Vocational Studies, Muzaffarnagar Uttar Pradesh, India
*Corresponding author: Ravi Kumar, SD College of Pharmacy & Vocational Studies, Muzaffarnagar Uttar Pradesh, India, Tel: 07417464903; E-mail: [email protected].
Received Date: June 30, 2022
Published Date: July 11, 2023
Citation: Kumar R, et al. (2023). Role of Biomarkers in Pathogenesis and Updated Treatment of Motor Neuron Disease: A Review Article. Mathews J Neurol. 7(2):24.
Copyrights: Kumar R, et al. © (2023).
ABSTRACT
One of the most severe adult-onset neurodegenerative illnesses is motor neuron disease (MND), which most frequently manifests as amyotrophic lateral sclerosis (ALS). Significant advancements in patient care have been made over the past ten years, and scientific research has advanced quickly. As a result, rational medicines based on important pathogenic pathways now appear conceivable. With an average one-year delay between the onset of symptoms and diagnosis, ALS exhibits considerable heterogeneity in both its presentation and subsequent course of clinical development. Despite the fact that 50% of patients pass away within 3–4 years of the onset of symptoms, usually from respiratory failure, a sizable minority makes it to their second decade. Recent genetic studies have discovered disease-causing mutations in genes in several seemingly unrelated functional pathways, suggesting that motor neuron degeneration may need to be understood as a common final pathway with a variety of upstream causes, despite the fact that it appears to be a sporadic disorder for the majority of patients and lacks obvious environmental triggers. Due to the apparent aetiological and clinical heterogeneity, treatment investigations should investigate genetic as well as clinical classification and involve thorough biomarker analysis. Hexanucleotide repeat expansion in C9orf72, which accounts for 10% of all cases of ALS in the Western Hemisphere, is the most frequent mutation. This gene, along with a number of others, points to altered RNA processing and protein breakdown pathways as the primary etiology of ALS. The research of pre-symptomatic gene carriers is an essential new project because there is still a significant knowledge vacuum on how such fundamental mechanisms appear to work without visible deficit in the decades before to the emergence of symptoms.
Keywords: Frontotemporal Dementia, Neurodegeneration, Anterior Horn Cell, RNA, TDP-43, Autophagy, Protein Aggregation, Amyotrophic Lateral Sclerosis.
ABBREVIATIONS
MND: Motor Neuron Disease; ALS: Amyotrophic Lateral Sclerosis; UMNs: Upper Motor Neurons; LMNs: Lower Motor Neurons; FTD: Fronto-Temporal Dementia; EMG: Electromyography; GWAS: Genome-Wide Association Studies; SNP: Single Nucleotide Polymorphism, MRI: Magnetic Resonance Imaging.
INTRODUCTION
Upper motor neurons (UMNs, such as the Betz cells of the motor cortex) and lower motor neurons (LMNs, such as the anterior horn cells of the spinal cord and brainstem nuclei) are lost in motor neurone disease (MND), an adult-onset neurodegenerative condition. As a result of the observation that most patients exhibit both UMN degeneration of the lateral corticospinal tract and its cortical origins manifesting as gliosis, or hardening (sclerosis), and LMN-related loss of muscle as a result of denervation (amyotrophy), the terms MND and amyotrophic lateral sclerosis (ALS) are frequently used interchangeably. There is no reversible treatment for what often results in the catastrophic collapse of a motor system that had previously appeared to be operating normally, despite multiple therapeutic treatments [1].
The notion that ALS affects only the motor system is no longer tenable. On histological examination, extra-motor cerebral pathology is frequently seen, even if it may not always be clinically evident, with a preference for the prefrontal, frontal [2], and temporal cortices. Up to 15% of patients have overt fronto-temporal dementia (FTD), which is linked with varying dysexecutive dysfunction, behavioral instability, and other symptoms. The idea that ALS and FTD are the extremes of a spectrum is now supported by a multitude of clinical and neuropathological evidence [3].
CLINICAL FEATURES AND DIAGNOSIS
Progressive motor weakening without sensory disruption is the ALS clinical characteristic.
A combination of UMN and LMN involvement typically causes loss of motor function, and since both are typically present on examination, there aren't many plausible mimic illnesses. Although it is rare for a reversible disease to be missed in this environment, rare 'pure' LMN or UMN cases, which are often also more slowly progressing, present the biggest diagnostic challenge. When patients are denied the proper support due to delayed diagnostics, it causes them great distress [4].
The symptom of fatality onset is a notable characteristic of ALS. Patients initially have a weak leg (60%) or dysarthria due to bulbar dysfunction (30%). Also common presentations are FTD or respiratory weakness (both 5%). The flail arm or "man-in-a-barrel" condition, which involves bilateral proximal and often LMN-predominant arm weakness, and the flail leg or "pseudopolyneuritic" syndrome, both of which typically advance significantly more slowly, are two distinctive spatially isolated forms of ALS that may be identified [5]. Although bulbar-onset ALS is frequently linked to a more rapid progression of symptoms, a subgroup, typically elderly women, may experience rapid cortico-bulbar involvement (UMN-predominant, with severe emotionality) that is isolated for many months, occasionally years, prior to the limbs weakening. Patients with bulgar-onset are commonly misreferred to TIA or ENT clinics, and over interpreting incidental spondylotic illness in those with limb weakness can lead to unneeded spine surgery [6].
As ALS advances, weakness gradually spreads to other bodily regions. If cognitive impairment does emerge, it usually does so early in the course of the disease, frequently entails behavioural changes, and is normally linked to more quickly progressing muscular weakness. Up to 50% of patients have a more modest dysexecutive condition, which does not, clearly, worsen as quickly as motor weakness does [7].
Ventilatory failure that occurs gradually is the usual cause of death in ALS. Even in those with severe bulbar weakness, choking is a rare cause of mortality, thus patients should be especially reassured of this.
A history of progressive motor dysfunction, such as weakness and loss of dexterity, and the presence of a combination of UMN and LMN signs are used to make the clinical diagnosis of ALS. Sometimes it can be challenging to elicit UMN signals. The electromyography (EMG) procedure can be used to show LMN enervation [8]. EMG, however, is only 60% sensitive; hence it shouldn't be used as a diagnostic or required inquiry. The El Escorial criteria, which link diagnostic certainty to the number of body regions affected clinically or neurophysiologically4 and categorise ALS as "possible" or "probable," have formalized the diagnostic process in the research setting. The Awaji criteria have improved the neurophysiological components of the El Escorial criteria, which probably increase their sensitivity, but they are still extremely strict in clinical practice. The objective of clinical evaluation is to make a reliable diagnosis as well as predict the course of each patient's disease to help plan the timing of therapies like gastroctomy or non-invasive ventilation. These achievements have served as the foundation for initiatives aiming at illness "staging" that could be advantageous in clinical trials [9].
EPIDEMIOLOGY
Though research is still lacking, it is believed that ALS occurs all over the world, particularly in China, India, and Africa, where thorough epidemiology has not yet been done. The Japanese Kii Peninsula and the Pacific island of Guam have been identified as hotspots for the ALS Parkinsonism-Dementia complex. These regions are unusual, their aetiology is still poorly understood, and they are pathologically distinct. Incidence rates for ALS were determined to be 2.6 per 100,000 for women and 3.9 per 100,000 for males in the UK, with lifetime risks of 1 in 472 and 1 in 350, respectively [10]. Although cases of ALS due to genetic abnormalities in FUS are occasionally detected in teenagers and often progress aggressively, the disease is extremely rare in those under the age of 30. The chance of developing ALS rises after the age of 40 and peaks in the early 1970s, after which there is an inexplicable fall in occurrence. A favourable prognostic indicator is a younger age at symptom onset. There is significant prognostic variation in ALS despite the fact that population-based studies have shown a median survival of about 3–4 years after the onset of symptoms. Although no consistent environmental risk factor has been found, many doctors have the strong sense that people with ALS have higher-than-average levels of pre-morbid physical fitness and a lower BMI. A genetic profile that predisposes to athleticism yet is perhaps more tolerant to motor system degeneration in later life could be the cause of such association, according to one conceivable explanation [11].
GENETICS
Long believed to affect only the 5% of ALS cases with a family history compatible with Mendelian inheritance, a genetic component to ALS was thought to be considerable.
Small family sizes and incomplete gene penetrance are just two of the many factors that contributed to the underestimation of the hereditary component of ALS [12].
Only 2% of all ALS cases (one-fifth of 'familial' cases) could be genetically explained for nearly two decades due to SOD1 gene abnormalities. Now, autosomal dominant single gene disorders are used to explain 75% of cases with a family history of ALS or FTD. An intronic hexanucleotide repeat expansion in open reading frame 72 (C9orf72) of chromosome 9 is the one and only most frequent genetic cause. In population-based research, mutations in SOD1, TARDBP, and FUS occur in less than 10% of cases, while mutations in other genes are even less prevalent [13].
40% of familial ALS instances and 25% of familial FTD cases are attributed to C9orf72 expansions, which might result in ‘pure’ ALS or FTD in family members? Although the clinical presentations of non-C9orf72 linked ALS and FTD are virtually indistinguishable, there seems to be an excess of ALS with cognitive impairment. The functional ramifications of the pathological expansion's drop in C9orf72 mRNA levels and the function of the encoded protein are unknown. RNA toxicity-related function gain is currently preferred as a pathogenic mechanism [14].
Even in the absence of a family history, it is probable that all occurrences of ALS are at least partially genetically determined. A lot of optimism was placed in genome-wide association studies (GWAS) to help reveal the "missing heritability," but few other findings have been repeated, with the exception of a strong relationship with a single nucleotide polymorphism (SNP) in the gene UNC13A [15]. Despite not always having biological significance, identified SNPs can serve as markers for genetic variants with which they are in linkage disequilibrium. Despite making a sizable contribution to the genetic makeup of the at risk population as a whole, loci found in GWAS are probably going to have modest odds ratios, and it may be difficult to determine their functional importance in terms of disease etiology.
PATHOLOGY AND PATHOGENESIS
The neuropathological hallmark of ALS is unique. In the motor cortex, brain stem, and spinal cord, loss of motor neuron cell bodies can be accompanied by astrogliosis and a sometimes ferocious microglial response.
The ubiquitinated protein TDP-43 has distinctive cytoplasmic inclusions in some glial cells and the remaining neurons. The TARDBP gene encodes TDP-43, a DNA and RNA-binding protein with a variety of roles in pre-mRNA splicing, translation regulation, and transcription. The protein is removed from its usual nucleus site in afflicted cells in addition to creating potentially hazardous cytoplasmic inclusions, indicating that a nuclear loss of function may also play a role in pathogenesis [16]. 'TDP-43 proteinopathies' are now thought to account for the great majority of ALS cases as well as a sizable part of non-tau FTD cases. Important exceptions include ALS cases associated with SOD1 and FUS, where TDP-43 is normal but SOD1 and FUS are abnormally aggregated and ubiquitinated [17].
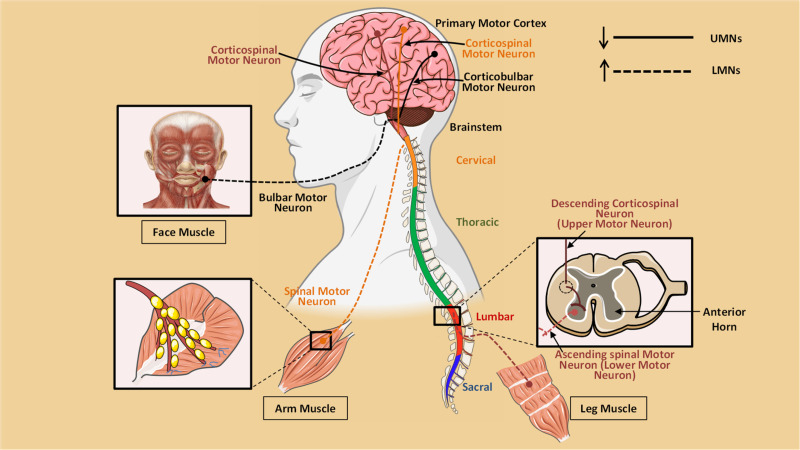
Figure 1. Pathophysiology of Motor Neuron Disease.
Although TDP-43 disease is present in C9orf72 expansion carriers as well, there is a distinct TDP-43 negative pathology that is characterized by ubiquitinated inclusions that comprise aggregates of improperly translated dipeptide repeats produced from the hexanucleotide repeat expansion. The repeat-associated non-ATG-initiated (RAN) translational mechanism, which does not require a start codon, is hypothesized to be the method by which this intronic region might generate peptide. Understanding the link between dipeptide repeat deposition, TDP-43 pathology and the clinical phenomenology is a priority [18]. These inclusions are most obvious in the cerebellum and hippocampus.
Since about 20 years ago, transgenic SOD1-overexpressed mouse models have been used to study ALS. With increased worry over the lack of therapeutic translation, this has led to a variety of putative pathogenic pathways, including loss of axonal transport, oxidative stress, mitochondrial dysfunction, and glutamate-mediated excitotoxicity. The identification of TARDBP and FUS mutations, as well as the knowledge that both of these RNA-binding proteins are related, has narrowed the emphasis of the investigation into upstream illness processes to RNA metabolism. Several more ALS-related genes, as well as those altered in other motor neuron illnesses that are not ALS, like spinal muscular atrophy, has roles in RNA metabolism. Therefore, a general theory is that changes have been made to RNA metabolism at the level of mRNA splicing, transport, or translation control. This might be caused by cytosolic or nuclear TDP-43 or FUS aggregation, loss, or both. RNA-binding protein homeostasis may be impacted if C9 or f72 has an aberrant hexanucleotide repeat [19].
Affected mRNA splicing results from both the overexpression of disease-causing TARDBP mutations and the loss of TDP-43 from mouse brain. Unusual protein aggregation is a key component of pathogenesis. Although ubiquitinated, intraneuronal inclusions are also connected to autophagy via p62, a protein that targets ubiquitinated payloads for removal. Both the ubiquitin-proteasome system (UPS) and autophagy are essential for cellular survival, and it's probable that in ALS, either because of an excess of aggregating protein or because the protein degradation machinery is fundamentally broken down, these systems are overloaded [20]. Gene alterations that are directly connected to mechanisms for protein degradation are the root cause of a number of unusual forms of ALS. These include UBQLN2, which codes for the UPS component ubiquilin 2, SQSTM1, which codes for p62, VCP, which codes for the protein that binds polyubiquitylated proteins and contains valosin, as well as others like OPTN and FIG4 [21].
BIOMARKERS
Biomarkers are required that could speed up diagnosis, enhance prognostic classification, and monitor therapy response. Trials presently use the revised ALS Functional Rating Score (ALSFRS-R) change in slope or survival as the primary outcome, both of which lack sensitivity. Though they require confirmation in sizable cohorts, CSF neurofilaments, TDP-43, and neuroinflammatory chemicals may reflect aspects of illness development and progression. Quantitative LMN biomarker possibilities are available in neurophysiology, such as electrical impedance myography and Motor Unit Number Estimation (MUNE) [22]. Transcranial magnetic stimulation-measured cortical hyperexcitability shows promising specificity for ALS.23 More sophisticated quantitative applications have evolved that are setting the standard for defining a systems-level biomarker signature, even though regular clinical MRI is commonly utilized to rule out structural lesions in the differential diagnosis of ALS. Across the spectrum of phenotypes, diffusion tensor imaging measurements of white matter tract integrity and voxel- and surface-based morphometry of the cortex have showed promise [23].
Consistent findings include loss of corpus callosum integrity, corticospinal tract thinning, and primary motor brain atrophy. [30] The so-called resting-state networks can be examined using functional MRI to detect alterations in functional connectivity that could be pathologically related to structural changes. It is hoped that a combination of markers would be successful even if no single neuroimaging result has yet made it possible to differentiate between disease states at the level of the person [24].
THERAPY
Disappointingly, Riluzole, which is believed to have a generally anti-glutamatergic mode of action, is the only medication to indicate survival benefit in human ALS. In a clinical trial context, it just slightly increased mean survival from 12 to 15 months [25].
On the basis of animal models of spinal cord injuries, new therapeutic approaches include those aimed at enhancing muscular performance. One such approach is the use of anti NOGO-A antibodies, which are thought to promote axonal development. The creation of multidisciplinary teams within specialized clinics, led by neurologists with expertise in ALS and supported by a specialist nurse, occupational therapists, speech therapists [26], dieticians, and physiotherapists, as well as links to gastroenterology and respiratory teams, has had a much greater impact on patients' quality of life. Nutritional therapy together with prompt gastrostomy is important interventions to take into account for maintaining patients with dysphagia's quality of life. Considerable survival as well as considerable quality-of-life advantages is provided by non-invasive ventilation. Patients rarely choose for invasive tracheostomy since it does not lessen the inevitable loss of limb function or the possibility of cognitive impairment. Palliative care services are frequently closely involved in the management of end-of-life in people with ALS [27].
FUTURE DEVELOPMENTS
The acknowledgment of cognition as the "third space" in addition to the conventional UMN and LMN pathology in ALS has led to the development of new pathogenic insights.
Since ALS and FTD are now recognized as spectrum disorders, the idea of selective susceptibility is expanded to include neural networks rather than specific cell types. The idea that harmful prion-like proteins may aid in the spread of disease across adjacent regions is currently under active consideration at the systems level [28].
This theory proposes that SOD1, TDP-43, and FUS, key ALS proteins, initiate foci of aggregation that spread from cell to cell via permissive templating, either from neuron to neuron or via glial cells. The concept of "spread" of disease is intimately related to the paradox of selective vulnerability in ALS, and it is an interesting fit to the clinical finding that disease appears to spread from a central point of onset to a nearby area [29]. It is unclear why some motor neuron subpopulations, such as those feeding the extraocular muscles, are noticeably resistant. These types of motor neurons differ physiologically, and distinctions in the transcriptome have started to appear. These developments may help us better understand the molecular basis of neuronal vulnerability [30,31].
It is becoming more and more obvious that ALS is a syndrome with a diverse etiology. Within the next ten years, the whole genetic contribution to this may be clarified thanks to the ability of current sequencing tools. This ought to result in the creation of enhanced disease models. The difficulty will be to mimic the broader consequences of biological aging in a realistic time frame. Currently, TDP-43, rather than SOD1-based rodent models, may better capture the majority of human ALS. Induction of pluripotent stem cells from patient fibroblasts can be used to create motor neuron-like cells, which have a higher relevance to disease than cell lines that over express cDNA constructs. It is possible that it will serve as a platform for high-throughput drug testing [32].
CONCLUSION
In this study, various biomarkers and have 2 main recently available drugs that possess potent pharmacological action. All neurological disorder like as ALS, UMND, LMND, FTD etc. are affect the neurons that control progression of signals and provide strength to muscular system. The ubiquitinated protein TDP-43 play a main role in the generation of neuronal defects and promotes to neurological disorders. According to previous research, mutation in TARDBP and FUS genes are also generate muscles atrophy and degeneration of neurons.
ACKNOWLEDGEMENTS
The authors thanks to Dr. Arvind Kumar, Director of SD College of Pharmacy & Vocational Studies, Muzaffarnagar U. P. India- 251001, for his all-time valuable cooperation.
FUNDING
No funding of any kind was received from any organization.
AUTHORSHIP
All named authors meet the journal's criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
CONFLICT OF INTEREST
All authors declare that they do not have conflict of interest.
REFERENCES
- Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. (2011). Amyotrophic lateral sclerosis. Lancet. 377(9769):942-955.
- Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S, et al. (2012). The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. 83(1):102-108.
- Turner MR, Talbot K. (2013). Mimics and chameleons in motor neurone disease. Pract Neurol. 13(3): 153-164.
- Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. (2000). El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 1(5):293-299.
- de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, et al. (2008). Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 119(3):497-503.
- Costa J, Swash M, de Carvalho M. (2012). Awaji criteria for the diagnosis of amyotrophic lateral sclerosis:a systematic review. Arch Neurol. 69(11):1410-1416.
- Roche JC, Rojas-Garcia R, Scott KM, Scotton W, Ellis CE, Burman R, et al. (2012). A proposed staging system for amyotrophic lateral sclerosis. Brain. 135(Pt 3):847-852.
- Beghi E, Logroscino G, Chiò A, Hardiman O, Mitchell D, Swingler R, et al. (2006). The epidemiology of ALS and the role of population-based registries. Biochim Biophys Acta. 1762(11-12):1150-1157.
- Alonso A, Logroscino G, Jick SS, Hernán MA. (2009. Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. Eur J Neurol. 16(6):745-751.
- Bäumer D, Hilton D, Paine SM, Turner MR, Lowe J, Talbot K, et al. (2010). Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology. 75(7):611-618.
- Turner MR. (2013). Increased premorbid physical activity and amyotrophic lateral sclerosis: born to run rather than run to death, or a seductive myth? J Neurol Neurosurg Psychiatry. 84(9):947.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 72(2):245-256.
- van Blitterswijk M, DeJesus-Hernandez M, Rademakers R. (2012). How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr Opin Neurol. 25(6):689-700.
- Andersen PM, Al-Chalabi A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 7(11):603-615.
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11(4):323-330.
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 314(5796):130-133.
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 339(6125):1335-1338.
- Turner MR, Bowser R, Bruijn L, Dupuis L, Ludolph A, McGrath M, et al. (2013). Mechanisms, models and biomarkers in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 14 Suppl 1(0 1):19-32.
- Bäumer D, Ansorge O, Almeida M, Talbot K. (2010). The role of RNA processing in the pathogenesis of motor neuron degeneration. Expert Rev Mol Med. 12:e21.
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 14(4):459-68.
- Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. (2013). Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology. 81(14):1222-1225.
- Turner MR, Kiernan MC, Leigh PN, Talbot K. (2009). Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. 8(1):94-109.
- Vucic S, Cheah BC, Yiannikas C, Kiernan MC. (2011). Cortical excitability distinguishes ALS from mimic disorders. Clin Neurophysiol. 122(9):1860-1866.
- Turner MR, Agosta F, Bede P, Govind V, Lulé D, Verstraete E. (2012). Neuroimaging in amyotrophic lateral sclerosis. Biomark Med. 6(3):319-337.
- Hardiman O, van den Berg LH, Kiernan MC. (2011). Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 7(11):639-649.
- Eisen A, Turner MR. (2013). Does variation in neurodegenerative disease susceptibility and phenotype reflect cerebral differences at the network level? Amyotroph Lateral Scler Frontotemporal Degener. 14(7-8):487-493.
- Polymenidou M, Cleveland DW. (2011). The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 147(3):498-508.
- Ravits JM, La Spada AR. (2009). ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 73(10):805-811.
- Brockington A, Ning K, Heath PR, Wood E, Kirby J, Fusi N, et al. (2013). Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 125(1):95-109.
- Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, et al. (2012). Mutant induced pluripotent stem cell lines recapitulate aspects of TDP43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci U S A. 109(15):5803-5808.
- Turner MR, Kiernan MC. (2012). Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotroph Lateral Scler. 13(3):245-250.
- Benatar M, Wuu J. (2012). Presymptomatic studies in ALS: rationale, challenges, and approach. Neurology. 79(16):1732-1739.