Information Links
Related Conferences
Previous Issues Volume 5, Issue 1 - 2021
Reticular Dysgenesis: Fiction or Reality in Morocco
Tshimanga S1*, Fouad A1, Fakiri K1, Rada N1, Draiss G1, Edmou B2, Bouskraoui M1
1Pediatrics Department, Mohammed VI University Hospital, Marrakesh, Morocco
2Immunology Department, Mohammed VI University Hospital, Marrakesh, Morocco
*Corresponding Author: Stanislas Tshimanga, Pediatrics Department, Mohammed VI University Hospital, PB 2360, Ibn Sina Avenue, Marrakesh, Morocco, Tel/Fax: +212524300700; E-mail: [email protected].
Received Date: February 03, 2021
Published Date: March 13, 2021
Citation: Tshimanga S, Fouad A, Fakiri K, Rada N, Draiss G, Edmou B. (2021). Reticular Dysgenesis: Fiction or Reality in Morocco. Mathews J Immunol Allergy. (5)1:12.
Copyrights: Tshimanga S, et al. © 2021. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
ABSTRACT
Severe combined immune deficits (SCID) are a group of rare yet the severest forms of primitive immune deficits. Reticular dysgenesis (RD) is a rare form of SCID characterized by lack of circulating T lymphocytes, severe congenital neutropenia, and sensorineural deafness. Mutations of the gene coding for adenylate kinase2 (AK2) have been identified as the molecular basis, and treatment consists in bone marrow transplantation. In this article, we report a case of reticular dysgenesis, a very rare condition in Morocco, and a review of the literature.
KEYWORDS: Adenylate kinase2; Reticular dysgenesis; Severe combined immunodeficiency
INTRODUCTION
Severe combined immune deficits (SCID) are a group of rare yet the severest forms of primitive immune deficits. They are hereditary anomalies characterized by a blocked differentiation of the T lymphoid lineage as well as defects of other hematopoietic lineages [1-3]. Discovered for over half a century now, reticular dysgenesis (RD) is a rare form of SCID characterized by lack of circulating T lymphocytes, severe congenital neutropenia, and sensorineural deafness. Mutations of the gene coding for adenylate kinase 2 (AK2) have been identified as the molecular basis, and treatment consists in bone marrow transplantation [4-9]. In this article, we reported a rarely described case in Morocco. Describe more about the disease, symptoms, and also describe AK2 gene responsible for this condition.
CASE REPORT
A 3-month-old infant born to first-degree consanguineous relatives was hospitalized for unprobed acute fever with a history of ecthyma gangrenosum and an episode of urinary infection without urinary tract malformation. The physical examination of the patient revealed a fever of 39.3°C, satisfactory staturo-ponderal development, with normal hemodynamic and respiratory functions, a urine test strip positive for nitrite, and oro-pharyngeal candidosis. There was evidence of auditory impairment. The otoacoustic emission was negative on both sides and the ophthalmological examination showed no peculiarities.
CLINICAL AND BIOLOGICAL SUMMARY OF THE CASE
|
Clinical |
Biological |
|
Age: 3 months |
Blood count: leukopenia (1460 cells / mm3), neutropenia (290 cells / mm3), lymphopenia (460 cells / mm3), Anemia (hemoglobin: 8.8 g/dl), MCV: 75, MCHC: 25, platelets count (154,000 cells / mm3). |
|
first-degree consanguineous relatives |
CRP: 11 mg/l |
|
History: inguinal ecthyma gangrenosum (Figure 1) and an episode of urinary infection |
blood urea: 0.22 g/l and serum creatinine: 2.4 mg/l |
|
Acute fever: 39,3°C Urine test strip positive for nitrite |
Blood culture: sterile, E. coli urinary infection on urine culture |
|
Satisfactory staturo-ponderal development |
HIV serology: negative |
|
Oro-pharyngeal candidosis |
Immunophenotyping of lymphocyte subpopulations: CD3 T: 188 / mm3, CD4 T: 10 / mm3, CD8 T: 10 / mm3 B CD19: 0 / mm3, NK CD16 / CD56: 22 / mm3. |
|
Ophthalmological examination: no peculiarities
|
Total immunoglobulins G: 0.852 g/l Ig M: 0.178 g/l Ig E and Ig A: No reagents. |
|
Otoacousticemission: negative |
CMV serology (IgM and IgG): positive CMV viral load: 12146 copies/ml. |
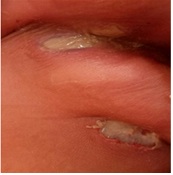
Figure 1: Inguinal Ecthyma gangrenosum.
Renal ultrasound revealed no abnormalities and the chest X-ray revealed no thymic shadow (Figure 2).
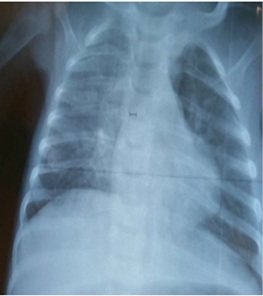
Figure 2: Chest X-ray: absence of thymic shadow.
In view of the above we concluded on a urinary tract infection (acute pyelonephritis) on severe combined immunodeficiency (SCID) complicated by systemic CME infection. Genetic consultation aimed at finding mutations in the gene coding adenylate kinase 2 was requested but not done for lack of necessary equipment.
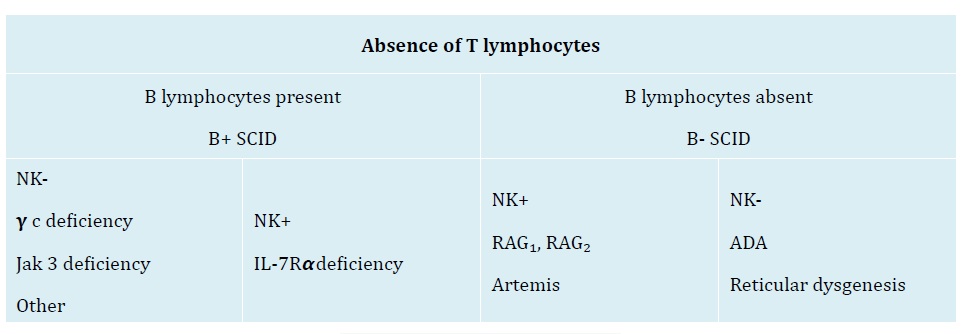
Figure 3: Classification of SCID [3].
The treatment included: cefotaxim 100mg/kg/day for the urinary infection, gancyclovir 5mg/kg/day for the systemic CMV infection, IV immunoglobulin 0,8g/kg/day every three weeks, while awaiting bone marrow transplant.
The progression was marked by death at home of the infant after the infectious episode duly treated successfully.
DISCUSSION
Severe Combined Immune Deficiency (SCID) is a group of rare primary immundeficiencies (PID) of the severest forms. Variable deficits in T, B, NK and other hematopoietic lineages are observed [1-3].
Primary immunodeficiencies are rare diseases: 1 in 500 to 1 in 500 000 in the general population (with considerable variations from one country to another) [10]. Hence the need for studies across different countries to improve our knowledge of PID, and to establish harmonized guidelines for better diagnostic and therapeutic management.
Reticular dysgenesis (RD) is perhaps the most severe type of SCID but extremely rare, comprising only 1–3% of SCID cases [11,12].
With a proportion of less than 2%, RD is a very rare form of SCID and since its first description in 20 patients, it has been reported in small series or in individual reports [12].
In addition to the classic combination of SCID T-B-NK- and agranulocytosis, patients with RD have severe sensorineural hearing loss [13-15], as seen in our case.
Manfred H. et al found in a transnational study that male and female children were affected by RD, and consanguinity was found in several families [8]. This was similar to our case.
Our patient had a history of Pseudomonas aeroginosa ecthyma gangrenosum. Pseudomonas aeroginosa is a ubiquitous gram-negative bacillus characterized by its greenish color and “grape-like” odor, and is the cause of potentially severe dermatoses such as ecthyma gangrenosum that indicates immunodepression or reveals sepsis, especially in children [16-18]. Also, Manfred H. et al. in their study found infectious agents including Pseudomonas aeroginosa isolated from blood cultures in 9 out of 19 patients with signs of sepsis [19].
Cosar H, et al. in a RD case study in a pre-term newborn found lymphopenia and neutropenia in the routine checkup [20]. The same finding was made by both Weinberg AG, et al. [21] and also in our study.
In 2009, mutations in the gene coding adenylate kinase2 (AK2) were identified as the molecular basis of the disease by two independent groups [14,15]. Reticular dysgenesis is caused by mutation in the encoding adenylate kinase2 (AK2), a mitochondrial protein important for regulating intracellular levels of adenosine diphosphate and maintaining mitochondrial membrane potential. A similar function is also mediated by the cytoplasmic enzyme AK1. While most tissues express both AK1 and AK2 enzymes, neutrophils, T lymphocytes and cells of striavascularis in the inner ear uniquely express AK2, thus explaining the RD phenotype [14, 15].
AK2 is critical to the control of energy metabolism. It regulates intracellular ATP levels by catalyzing the reaction ATP + AMP D 2ADP contributes to ~60% of the ADP flux in the mitochondrial matrix [15]. Since AK2, is the only adenylate kinase expressed in the bone marrow; its absence is expected to severely disrupt mitochondrial function and oxidative phosphorylation leading to a profound block in lymphoid and myeloid cell differentiation [22-24].
Different AKs set the adenine nucleotide ratio in different intracellular compartments. In particular, AK2 is expressed in the mitochondrial intermembrane space where it regulates the concentration of mono- and diphosphorylated adenine nucleotides [25,26].
Thus Manfred H, et al [19] reported 22 different mutations of AK2 during their study of 30 patients from 27 different families. In two of the patients, no sample was available for retrospective genetic analysis. With respect to our case, this analysis was not carried out for lack of necessary equipment (Figure 4).
.jpg)
Figure 4: Localization and function of human adenylate kinase 2.
In addition to the poor profile of T cells, the bone marrow normally produces erythrocytes and platelets. However, the patient may develop secondary anemia as a result of infectious processes. [27,28]. This was the case of our patient who later developed anemia with a normal platelet count.
Unlike children with other syndromes who have congenital neutropenia, those with RD fail to respond to granulocyte colony-stimulating factor, resulting in early death from infection unless they undergo successful transplantation [4-9].
Haematopoietic stem cell transplantation is currently the only option to correct the immunological and haematological phenotype of RD [4-9, 14,19].
CONCLUSION
Knowledge of reticular dysgenesis is invaluable in that the survival of patients directly depends on the speed with which diagnosis is made and treatment started.
CONFLICT OF INTEREST
None.
CONSENT FOR PUBLICATION
A written informed consent was obtained from all patients when they were enrolled.
REFERENCES
- Fischer A, Le Deist F, Hacein-Bey-Abina S, André-Schmutz I, de Saint Basile G, et al. (2005). Severe combined immunodeficiency. A model disease for molecular immunology and therapy. Rev Immunol. 203(1):98-109. http://dx.doi.org/10.1111/j.0105-2896.2005.00223.x].
- Aloj G, Giardino G, Valentino L, Maio F, Gallo V, et al. (2012). Severe combined immunodeficiencies: new and old scenarios. Int Rev Immunol. 31(1): 43-65. [http://dx.doi.org/10.3109/08830185.2011.644607].
- Bertrand Y, Baleydier F. (2010). Diagnostic d’un déficit immunitaire primitive de l’enfant. Revue francophone des laboratoires. 40(424):53-58.
- Levinsky R, Tiedman K. (1983). Successful bone marrow transplantation for reticular dysgenesis. Lancet. 1(8326): 671-2. [http://dx.doi.org/10.1016/s0140-6736(83)91968-2].
- Fischer A, Landais P, Friedrich W, Morgan G, et al. (1990). European experience of bone marrow transplantation for severe combined immunodeficiency. Lancet. 336(8719): 850-4. [http://dx.doi.org/10.1016/0140-6736(90)92348-1].
- De Santes KB, Lai SS, Cowan MJ. (1996). Haploidentical bone marrow tranplants for two patients with reticular dysgenesis. BMT. 17(6): 1171-3.
- Antoine C, Müller S, Cant A, cavazzana-Calvo M, Veys P, et al. (2003). Long-term survival and transplantation of haemopoietic sterm cells for immunodeficiencies: Report of the European experience 1968-99. Lancet. 361(9357): 553-560. [http://dx.doi.org/10.1016/s0140-6736(03)12513-5].
- Manfred H, Pannicke U, Hubert B, Schwarz K. (2018). Recent advances in understanding the pathogenesis and management of reticular dysgenesis. Br J Haematol. 180(5): 644-53. [http://dx.doi.org/1010.1111/bjh.15045].
- Ownby DR, Pizzo S, Blackmon L, Gall SA, Buckley RH. (1976). Severe combined immunodeficiency with leukopenia (reticular dysgenesis) in siblings: immunologic and histopathologic findings. J Pediatr. 89(3):382-7. [http://dx.doi.org/101016/s0022-3476(76)80532-x].
- De vries E. (2006). Patient-centred screening for primaryimmunodeficiency: amilti-stage diagnostic protocoldesigned for non-immunologists. Clinical and Experimental Immunology. 145(2):204-14. [http://dx.doi.org/10.1111/j.1365-2249.2006.03138.x].
- Stephan JL, Vlekova V, Le Deist F, Blanche S, Donadieu J, et al. (1993). Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr. 123(4):564-72. [http://dx.doi.org/10.1016/10022-3476(05)80951-5].
- Och HD, Edvard Smith CI, Puck JM. (2014). Primary Immunodeficiency Diseases: A Molecular and Cellular approach. Oxford University Press. 269-273.
- Small TN, Wall DA, Kurtzberg J, Cowan MJ, O'Reilly RJ, et al. (1999). Association of reticular dysgenesis (thymic alymphoplasia and congenital aleukocytosis) with bilateral sensoneurol deafness. J Pediatr. 135(3):387-89. [http://dx.doi.org/10.1016/s0022-3476(99)70141-1].
- Lagresle-Peyrou C, Six EM, Picard C, Rieux-Laucat F, Michel V, et al. (2009). Human adénylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet. 41(1):106-11. [http://dx.doi.org/10.1038/ng.278].
- Pannick U, Honnig M, Hess I, Friesen C, Holzmann K, et al. (2009). Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adénylate kinase 2. Nat Genet. 41(1):101-5. [http://dx.doi.org/10.1038/ng.265].
- Morand A, Morand JJ. (2017). Pseudomonas aeroginosa en dermatologie. Annale de dermatologie et de vénérologie. 144(11):666-75.
- Carsuzaa F, Gissert O, Bove T, Terrier JP, et al. (2020). L’ecthyma gangréneux. Rec Med interne. 23(1):159.
- Boisseau M, Sarlangue J, Perel Y, Hehunstre JP, Taïeb A, et al. (1992). Perineal ecthyma gangrenosum in infancy and earl childhood: septicemic and nonsepticemicforms. J Am Acad Dermatol. 27(3):415-8. [http://dx.doi.org/10.1016/0190-9622(92)70210-7].
- Manfred H, Lagresle-Peyrou C, Pannicke U, Notarangelo LD, Pota F, et al. (2017). Reticular Dysgenesis; international survery on clinical presentation, transplantation, and Outcome. Blood. 129(21):2928-38. [http://dx.org/10.1182/blood-2016-11-745638].
- Cosar H, Kahramaner Z, Erdemir A, Kanik A, Turkoglu E, et al. (2010). Reticular Dysgenesis in preterm infant: A case Report. Pediatric Hematology and Oncology. 27(8): 646-9. [http://dx.org/10.3109/08880018.2010.509424].
- Weinberg AG, Rosenfeld CR, Manroe BL, Browne R. (1985). Neonatal blood cell count in health and disease. II. Values for lymphocytes, monocytes, and eosinophyles. J Pediatr. 106(3):462-6. [http://dx.doi.org/10.1016/s0022-3476(85)80681-8].
- Six E, Lagresle-Peyrou C, Susini S, De Chappedelaine C, N Sigrist, et al. (2015). AK2 deficiency compromises the mitochondrial energy metabolism required for differenciation of human neutrophil and lymphoid lineages. Cell Death Dis. 6(8):e1856. [http://dx.doi.org/10.1038/cddis.2015.211].
- Noma T. (2005). Dynamics of nucleotide metabolism as a supporter of life phenomena. The journal of Medical Investigation. 52(3-4):127-36. [http://dx.org/10.2153/jmi.52127].
- Noma T, Murakami R, Yamashiro Y, Fujisawa K, S Inouye, et al. (2000). cDNA cloning and chromosomal mapping of the gene encoding adenylate kinase2 from drosophila melanogaster. Biochimica and biophysica Acta. 1490(1-2):109-14. [http://dx.doi.org/10.1016/s0167-4781(99)00223-7].
- Dzeja P and Terzic A. (2003). Phosphotransfer networks and cellular energetics. J Exp Biol. 206(12):2039-47.
- Dzeja P, Shung C, Terzic A. (2007). Integration of adenylate kinase and glycolytic and clycogenolytic circuit in cellular anergetics. I Molecur System Bioenergetics. 265-301. [http://dx.doi.org/10.1002/9783527621095.ch8].
- De le Calle-Martin O, Badell I, Garcia A, Gelpi C, Casamitjana N, et al. (1997). B cells and monocytes are not developmentally affected in a case of reticular dysgenesis. Clin ExpImmunol. 110(3):392-6. [http://dx.doi.org/10.1046/j.1365-2249.1997.4341453.x].
- Espagnol T, Compte J, Alvarez C, Tallada N, Laverde R, et al. (1979). Reticular dysgenesis: report of two brothers. Clin ExpImmunol. 38(3):615-20. [PMID: 535190].