Previous Issues Volume 1, Issue 1 - 2017
Regenerative Therapy in Autosomal Recessive Polycystic Kidney Disease
Mohamed SA Mohamed1
1University of Cologne, Deutz-Kalker Str. 118, 50679, Cologne, Germany.
Corresponding Author:Mohamed SA Mohamed, University of Cologne, Deutz-Kalker Str. 118, 50679, Cologne, Germany,Tel: +49 221 8275-2456; Email: [email protected]
Received Date: 13 Jun 2016
Accepted Date: 17 Jun 2016
Published Date: 20 Jun 2016
Copyright © 2016 Mohamed MSA
Citation: Mohamed MSA. (2016). Regenerative Therapy in Autosomal Recessive Polycystic Kidney Disease. Mathews J Surgery. 1(1): 002.
ABSTRACT
Autosomal recessive polycystic kidney disease (ARPKD) is a developmental disease that results from fibrocystin loss of function mutation. It affects mainly renal collecting ducts and biliary system. The exact mechanism of how fibrocystin mutation results in the disease manifestations is not known as the exact functions of fibrocystin are not very well identified. One of the mechanisms involved in the development of ARPKD is the impaired eipthelial cell polarity, which renders the epithelium secretory rather than absorptive. This paper sheds the spot on a hypothesis that the impaired epithelial cell polarity seen in ARPKD might result from the impaired β1β2 Na+/K+ ATPase expression together with the impaired focal adhesion kinase (FAK) activation and function, with the possible involvement of the adhesion signalling in the development of those interactions. Restoration of the functions of FAK and β1 Na+/K+ATPase in the renal and biliary epithelia of ARPKD patients might be a future hope of recovery that could be achieved through the regenerative therapy, using stem cells that express myroslated FAK and β1 Na+/K+ ATPase subunit to slowdown or reverse cyst formation.
KEYWORDS
Arpkd; Fibrocystin; Fak; Na+/K+ ATPase; Stem Cell Therapy and Epithelial Cell Polarity.
INTRODUCTION
Sodium-potassium adenosine tri-phosphatase (Na+ /K+ ATPase) is an enzyme that present in the plasma membrane of all mammalian cells and works to pump Na+ out and K+ in against their concentration gradients, obtaining the required energy through the hydrolysis of ATP [1]. The transmembrane protein complex is composed of an a and a β subunit. The a subunit is the channel with ATPase activity, while β subunit has a regulatory function. Naturally occurring isoforms for every subunit include a1 & a2 and β1 & β2 [2]. While in action, the pump binds an ATP molecule and 3 intracellular Na+ ions. As the ATP is hydrolyzed, phosphorylation of the pump at a highly conserved aspartate residue occurs and subsequently ADP is released. This is followed by a conformational change that exposes the Na+ ions to the cellular outside. As the phosphorylated form of the pump has a low affinity for Na+ ions, this allows the extracellular Na+ release. Afterwards, the pump binds 2 extracellular K+ ions, which facilitates the de-phosphorylation of the pump, reverting it to its original conformational state, transporting the K+ ions to the cell interior. As the de-phosphorylated form of the pump has a low affinity for K+ ions, this allows the intracellular K+ release [1] (Figure 1).
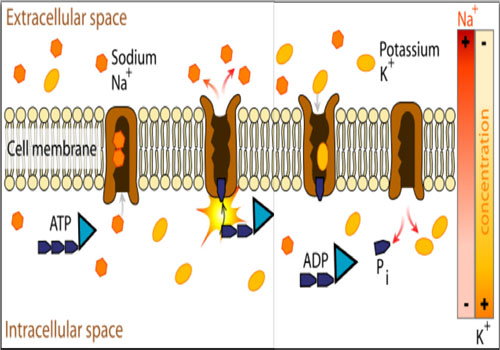
Figure 1: Diagrammatic representation of the mechanism of action of Na+ /K+ ATPase. Released for public use without copyrights by Lady-ofHats Mariana Ruiz Villarreal - Own work.
FUNCTIONS OF NA+ /K+ ATPASE
NNA+/K+ ATPase helps to maintain the resting membrane potential and effective transport across the cell membrane, and accordingly, regulates the cellular volume.
Resting Potential In order to maintain the physiological cell membrane potential, the intracellular concentrations of Na+ and K+ are kept under a physiological balance, where a low concentration of Na+ and high levels of K+ are favored. As the Na+ /K+ ATPase moves 3 Na+ out and 2 K+ in, thus, in total, removing one positive charge from the intracellular space, this creates a differential resting membrane potential between the cell membrane interior and exterior. Disturbance of this physiological balance, through the intracellular flow of Na+ , results in cell membrane depolarization [1].
Transport Export of sodium from the cell provides the driving force for several secondary active membrane transport proteins, which import glucose, amino acids, and other nutrients into the cell. In addition, the pump is involved in the creation of the transmembrane Na+ current that is important for many mechanisms of homeostasis [1].
Control of the cell volume As Na+ is the main extracellular cation that can diffuse intracellularly in response to its concentration gradient, which if happened would increase the intracellular osmotic pressure, leading to cell swelling and cell death. By keeping the active extracellular Na+ transport against its concentration gradient, Na+ /K+ ATPase plays an important role in the control of the cell volume [1].
Functioning as signal transducer While the a subunit is the channel with the ATPase activity, β subunit has regulatory and signaling functions. These functions involve the activation of the mitogen-activated protein kinase (MAPK) signal cascade and the activation of phospholipase C (PLC) and inositol triphosphate (IP3) receptor (IP3R) signalling [2, 3].
AUTOSOMAL RECESSIVE POLYCYSTIC KIDNEY DISEASE
Autosomal recessive polycystic kidney disease (ARPKD) is a developmental disease that affects 1/ 10000- 1/ 40000 live births worldwide. The disease manifestations usually starts in the neonatal period with enlarged echogenic kidneys. About 45% of infants have liver abnormalities, including hepato megaly, dilated intra-hepatic (and occasionally extra-hepatic) biliary ducts, at the initial presentation. Associated oligohydramnios can occur in a number of affected infants, resulting in pulmonary hypoplasia that could be the cause of death [4]. Up to 30% of the affected infants die in the neonatal period or within the first year of life because of the respiratory insufficiency and or the superimposed infections, while most of the survivors progress to end-stage renal disease (ESRD) in the first decade of life. With neonatal respiratory support and renal replacement therapies, the 10-year survival of ARPKD patients can reach 82%, while the 15-year survival is could reach 67%-79%. Other complications in older childhood or young adulthood are hepato-splenomegaly and portal hypertension [4]. ARPKD results from Fibrocystin loss of function mutations. Fibrocystin is encoded by pkhd1, an extremely large gene that comprises 86 coding exons. Fibrocystin is a large, transmembrane, receptor-like protein of 500 kDa, which is thought to be involved in the process of tubulogenesis and/or the maintenance of the duct lumen of the epithelium. It associates with the basal bodies and the primary cilia in renal epithelial cells (Figure 2) [5]. Pathogenesis of ARPKD involves three major processes; Proliferation of epithelial cells; Dilatation of the lumen; Secretion of fluid into the lumen. Accordingly, the epithelial cells of the renal collecting ducts, which line the growing cysts in ARPKD, secret fluid into cysts’ lumina (Figure 3) [6].
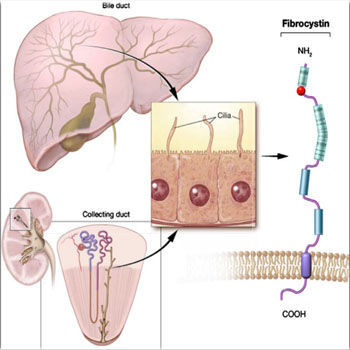
Figure 2: Diagrammatic representation of the ultra-structure of the fibrocystin (polycystin) molecule, which is expressed in the cilia of both the bile ducts of the liver and collecting ducts of the kidney. Citation; Pediatric Radiology© Springer-Verlag 2008, Vol.10, 1007.
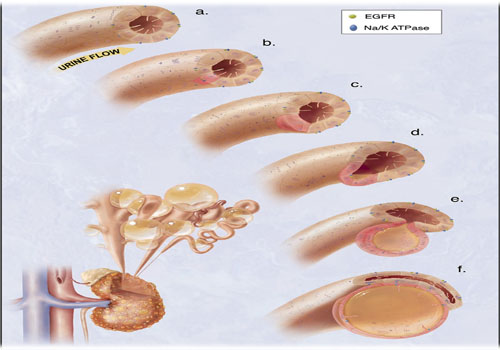
Figure 3: hypothetical model of the cystogenesis. “The illustration depicts mechanosensory function of renal tubular epithelial cilium. a. Each cilium plays an important role to transmit extracellular information, such as urine flow, into the cell. This message may provide critical signals to the cell regarding the direction of cell division along the tubule. b. Insults, such as genetic disorder or random mutation, will result in abnormal ciliary function to sense fluid movement. c. The functional abnormality in ciliary sensing may result in loss of planar cell polarity. d. Direction of cell division becomes randomized, resulting in increasing tubular diameter rather than tubular elongation. e. Budding of a cyst from the renal tubule and abnormal localizations of epidermal growth factor receptor (EGFR) and Na+ - K+ ATPase pump are typical characteristics of polycystic kidney. f. The cyst is eventually enlarged and isolated. Multiple cysts from the neighboring nephrons are illustrated on the bottom left corner”. Citation; Frontiers in Bioscience 2008, Vol.13, 4451-4466.
Much research work has been conducted for the investigation of the role of fibrocystin mutations in the development of the abnormal renal/ biliary epithelial cells proliferation/ apoptosis and tubular dilatation, which revealed the involvement of many pathways (Figure 4) [7-10]. However, till moment there is no clear explanation for how a fibrocystin mutation can be involved in the development of the intra-cystic fluid secretion.
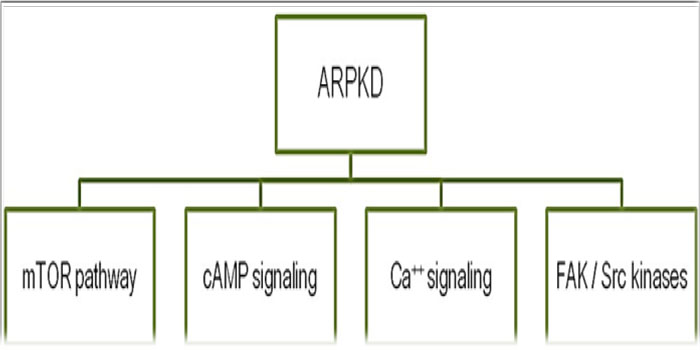
Figure 4: Pathways reported to be involved in the development of ARPKD.
During the development of ARPKD, the following findings have been observed;
• Loss of fibrocystin function leads to defective FAK activity [11]. • The expression of the β1 Na+ /K+ ATPase subunit is decreased for the sake of β2 subunit [2]. • Cellular polarization is disturbed and Na+ /K+ ATPase localizes to the apical rather than the lateral cellular borders which contributes to a major role in disease pathogenesis [12, 13].
ROLE OF NA+ /K+ ATPASE AND FAK
Under physiological conditions, both FAK and β1 Na+ /K+ ATPase subunit localize to the cellular adhesions, where both are suggested to be involved in controlling cellular polarization and intracellular signalling [11-13]. The localization of Na+ /K+ ATPase affects the physiological function of the duct epithelium. When Na+ /K+ ATPase localizes to the basolateral borders of the duct epithelial cells, while epithelial sodium channel (ENaC) localizes to the apical borders, such physiological localization results in the flow of Na+ from the lumen into the cytoplasm, which is followed by their pumping into the interstitial spaces and sub-epithelial tissue. Accordingly, water follows the higher osmolarity and the epithelium is absorptive [12]. In ARPKD, mislocalization of Na+ /K+ ATPase from the basolateral to the apical epithelial cellular borders has been reported (Figure 3), which coincided with a switch from the physiological β1 subunit expression to an enhanced expression of β2 subunit [12, 13].Accordingly, Na+ ions are secreted into the lumen and the water follows, which renders the epithelium secretory. Nevertheless, physiologically localized (basolateral) Na+ /K+ ATPase was found to be essential for the fusion of the multiple developing lumina into one lumen during renal tubulogenesis, as well as for the maintenance of normal tube diameter through maintaining normal fluid flow. Block of Na+ / K+ ATPase has been reported to result in the dilatation of the tubules with epithelial proliferation secondary to dilatation [12]. However, how Fibrocystin loss of function mutation can be involved in the process of Na+ /K+ ATPase mislocalization! During development, epithelial cellular borders cannot be marked into apical and basolateral due to the absence of tight junctions between adjacent cells, which coincides with the increased expression of β2 Na+ /K+ ATPase subunit. However, at late developmental stages, with the development of tight junctions, epithelial cells polarize into apical and baso- lateral borders, with Na+ /K+ ATPase localizes to the baso-lateral cellular borders under physiological conditions, which coincides with increased expression of β1 subunit [2]. β1 subunits were found to localize to tight junctions and focal adhesions, where it interacts with the adhesion signaling proteins [14,15].The cell adhesion molecule E-cadherin has been implicated in the development and the maintenance of the polarity of the epithelial cells. Transformed MDCK cells with reduced levels of Ecadherin and β1 Na+ /K+ ATPase subunit showed disturbed polarization and increased migration. Expression of E- cadherin in these cells, without re- expression of β1 Na+ /K+ ATPase subunit, fail to re-establish cellular polarization. While re- expression of both proteins resulted in re-establishment of polarity and reduced cell motility. Which remarks a very important role of β1 Na+ /K+ ATPase subunit in the regulation of cell polarization [15-17]. FAK is a very potent and important intracellular tyrosine kinase, which plays many important roles in adhesion signaling, cellular motility, cellular survival and growth. FAK interacts with Src tyrosine kinase and form a complex that exerts a group of synchronized tyrosine phosphorylation actions, which mediate many vital cellular processes. Phosphorylation of FAK at different tyrosine residues results in various activities. FAK phosphorylation at Y397 results in autophosphorylation and auto-activation [11, 18]. It was described that Fibrocystin mutation or knockdown leads to defective phosphorylation of FAK at Y397, resulting in inappropriate FAK activity [11]. Meanwhile, RNAi studies established that FAK is important for the polarized cell migration [17, 18]. FAK deficient fibroblasts fail to establish proper polarized migration even with increased Src expression. This role of FAK is mediated through signaling interactions with other cytoskeletal and focal adhesion proteins [18]. All those findings support the notion that FAK might be involved in regulation of cell polarity. Taken together, the decreased expression of β1 Na+ /K+ ATPase subunit, which is necessary for the E- cadherin mediated establishment of cell polarity, and decreased activation of FAK, which interacts with adhesion proteins and is essential for the establishment of the polarized cell migration, both together may explain how Fibrocystin loss of function mutation can result in the reversed epithelial cell polarity and the intra-cystic secretion in ARPKD. As Na+ /K+ ATPase has the ability to inhibit Src kinase activity, the increased Src expression may reflect defective Na+ /K+ ATPase and FAK signalling [19, 20]. Accordingly, the experimental proof of the above scenario could be favored if: • Defective expression of Fibrocystin was reflected on the FAK expression/function. • In the ARPKD cells, where the expression of β1 Na+ /K+ ATPase subunit is defective, Src expression is enhanced.
SOME SUPPORTIVE DATA
Three independent repeats of Fibrocystin knockdown in MDCK II cells were performed and FAK bands were assessed with immunoblotting (Figures 5 and 6). The results showed double fold increase of FAK in response to Fibrocystin knockdown. In addition, FAK bands were obviously denser in cells of ARPKD renal collecting tubules than in cells of age- matched human fetal renal collecting tubules (HFCT), as presented by Israeli et al[11] (Figure 7). This would prove the involvement of defective FAK activity in the development of ARPKD, as the defective expression of Fibrocystin resulted in the defective FAK signaling, which force the cell to induce it.
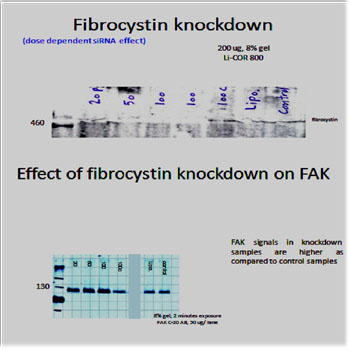
Figure 5: after optimization of the western blot protocol, the 500 kDa fibrocystin bands were detected and knockdown experiment was done using specific siRNA and lipofectamine reagent (Invitrogen). 0.5X 106 cells were seeded in 6 wells plate and incubated overnight till 70% confluency. The used siRNA concentrations for treatment of cells were 20, 50, 100 pmol/ ml, in addition to 100 pmol/ ml control non- specific siRNA treatment, lipofectamine only treatment and control non-treated cells. Cells were incubated after treatment for 48 hours before they were washed twice with cold PBS and scraped, on ice, in 0.5 ml RIPA buffer supplemented with proteases inhibitors. After sonofication, cellular extracts were centrifuged to get rid of debris and protein concentration of each sample was determined according the laboratory protocol. FAK bands were detected using FAK C-20 antibody (Santa Cruz). Analysis of the results was done using Licor system software. Dose dependent fibrocystin knockdown resulted in corresponding dose dependent FAK induction.
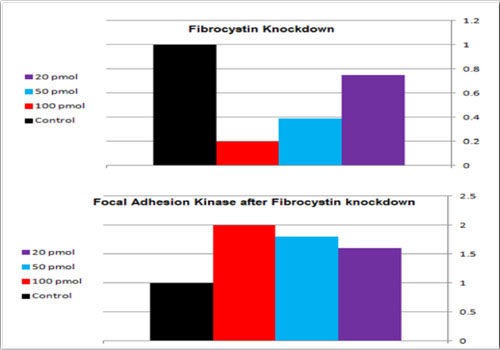
Figure 6: Effect of fibrocystin knockdown on FAK bands in MDCK II cells. With 100 pmol/ml siRNA, 80% fibrocystin knockdown was achieved, with corresponding double fold increase in FAK band density.Effect of fibrocystin knockdown on FAK bands in MDCK II cells. With 100 pmol/ml siRNA, 80% fibrocystin knockdown was achieved, with corresponding double fold increase in FAK band density.
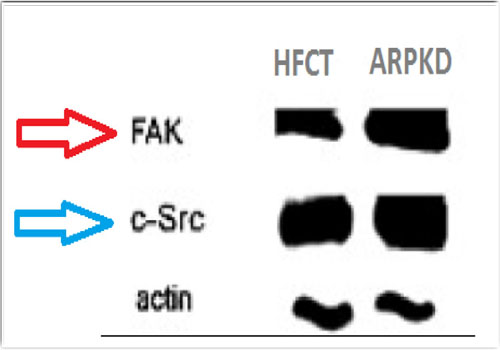
Figure 7: Increased densities of FAK and Src bands in ARPKD cells, in comparison to age matched human fetal collecting tubules (HFCT) cells. Citation; Am J Physiol Cell Physiol. 2010; 298 (4): C831-46.
Accordingly, it could be concluded that the impaired epithelial cellular polarity seen in ARPKD might result from the impaired β1 /β2 Na+/K+ ATPase expression together with the impaired FAK activation and function, with the possible involvement of the adhesion signalling in the development of those interactions. Restoration of the functions of FAK and β1 Na+ /K+ ATPase in the renal and biliary epithelia of ARPKD patients could be a future hope of recovery that could be achieved through the regenerative therapy, using stem cells that express myroslated FAK and β1 Na+ /K+ ATPase subunit to slowdown or reverse cyst formation.
RENAL VERSUS MSCS TRANSPLANTATION FOR ARPKD
Infants with ARPKD usually develop renal failure in very young age. Although renal transplantation might be a life-saving intervention, it could not be applied before the size of 10 Kg, in order to allow the hemodynamic capacity to reliably perfuse the graft and to limit the chance of graft thrombosis and acute tubular necrosis. In addition, this could be the minimal size that allows the peritoneal cavity to enclose the transplanted kidney without respiratory complications [21]. However, achieving the size of 10 Kg is not an easy target in ARPKD patients, as the growing polycystic kidneys compress the lungs and the stomach, and contribute to false weight gain. Moreover, ARPKD is usually a combined disease that affects the kidneys and the liver, which might necessitate combined liver and kidney transplantation [21]. This might raise the importance of an alternative regenerative therapy. Mesenchymal stem cells (MSCs) are multipotent stromal cells that can differentiate into a variety of cell types, such as fat, bone, and cartilage cells. Apart from the mesenchymal lineages, it has been reported that MSCs could also give rise to skeletal myocytes, cardiomyocytes, smooth muscle cells, and endothelial cells [22]. MSCs are well tolerated by the immune system, and have the ability to attenuate immunity through modulating T cell activation and proliferation, either by a direct cell-cell interaction or via soluble factors [23, 24]. This effect is independent on MHC matching as MSCs are characterized by low expression level of MHC I and II, and lacking of T cell co-stimulating molecules CD80 and CD86 [25]. To be able to achieve a regenerative therapeutic effect, migration and homing of MSCs are very important steps, which determine the effective usability of MSCs, as the cells must be delivered to the organ and the site of interest, to be able to differentiate into the particular cell type, and to replace or improve the healing of the addressed injury [26]. Accordingly, endovascular admistration of MSCs into the renal arteries might constitute a promising therapeutic strategy. Being tolerated by the immune system, allografting of MSCs from any person, regardless of the degree of MHC matching, might be applicable. However, it would still be better to obtain the used MSCs from the patient's relatives or a MHC matching person. Nevertheless, considering the fact that MSCs gradually lose their differentiation ability with aging, attention should be paid to the timing of MSCs transplantation, where 48 hours culture and harvest upon 90% confluence would allow good expression levels of the transfected myroslated FAK and β1 Na+ /K+ ATPase subunit [27, 28]. Nevertheless, cellular biology, molecular biology as well as clinical studies should be conducted to roll out the possibility of involvement of MSCs therapy in cancer development, which is not fully clear at the moment. A considerable number of studies has confirmed the tropism of MSCs to cancer. While some studies notified the immune-modulatory effect of MSCs to be a potential for cancer promotion, other studies suggested an anti-cancer role of MSCs. This controversy was reviewed by Hiroshi Yagi and Yuko Kitagawa, however, more understanding and safety studies should precede the application of MSCs therapy in the clinical practice [29].
CONFLICTS OF INTEREST
The experiments performed for the testing of the hypothesis were performed in the laboratory of pediatrics nephrology, Hanover medical school, where all the funds were supplied by the director of the department. The intellectual properties included in this manuscript belong solely to the author. The author welcomes funding cooperation for further testing of the hypothesis.
REFERENCES