Previous Issues Volume 3, Issue 1 - 2018
Molecular Biology and Cellular Signaling Pathways in Glioblastoma
Nikolaos Andreas Chrysanthakopoulos 1, Panagiotis Andreas Chrysanthakopoulos 2
1Dental Surgeon (DDSc), Department of Pathological Anatomy, University of Athens, Athens, Greece.
2Colonel-Neurosurgeon (MD), Director of Neurosurgery Clinic, 'NIMTS', Military Hospital, Athens, Greece.
Corresponding Author: Nikolaos Andreas Chrysanthakopoulos, Dental Surgeon (DDSc), Oncologist, Resident in Maxillofacial and Oral Surgery, 401 General Military Hospital of Athens, Athens, Greece, Tel: +30-2610-225288; E-Mail: nikolaos_c@hotmail
Received Date: 22 Sep 2018 Accepted Date: 01 Oct 2018 Published Date: 05 Oct 2018 Copyright © 2018 Chrysanthakopoulos NA
Citation: Chrysanthakopoulos NA and Chrysanthakopoulos PA. (2018). Molecular Biology and Cellular Signaling Pathways in Glioblastoma. Mathews J Neurol. 3(1): 012
ABSTRACT
Glioblastoma, also known as glioblastoma multiforme (GBM) or diffuse astrocytoma, WHO grade IV is the most aggressive and frequent primary brain cancer characterized by extremely poor prognosis despite developments in molecular Biology and genetics and new anti-neoplasmatic treatments and shows a great morphological and genetical heterogeneity.
Cancer pathogenesis characterized by the accumulations of molecular abnormalities and abnormalities in cellular signaling pathways that can lead to the known hallmarks of cancer:sustaining proliferative signaling due to mutations in proto-oncogenes, evading growth suppressors due to mutations affecting the tumor suppressor genes, enabling replicative immortality due to telomerase activation, resisting cell death due to up-regulation of anti-apoptotic or down regulation of pre-apoptotic molecules, inducing angiogenesis, activating tissue invasion and metastasis. GBM's molecular biology and pathogenesis are still complicated and in several aspects remain still unknown. It is characterized by rapid infiltrating growth, and microvascular proliferation and/or by expressing tissue necrosis. Its development could be as a primary tumor or as a secondary due to a malignant transformation that can arise from a lower grade brain tumor and/or with the isocitrate dehydrogenase (IDH) gene mutation. Previous and recent researches have recorded approximately 20 mutated genes that are implicated in GBM pathogenesis that are promoted by genomic instability and alternative pathways.
KEYWORDS
Glioblastoma; Gliomas; Signaling Pathways; Genomic.
INTRODUCTION
Glioblastoma, also known as glioblastoma multiform (GBM) or diffuse astrocytoma, WHO grade IV is the most aggressive and frequent primary brain cancer characterized by extremely poor prognosis shows a great morphological and genetical heterogeneity. GBM can be presented as a primary or as a secondary tumor as a result of a malignant transformation from a lower grade brain tumor and/or with mutation in the gene of isocitrate dehydrogenase (IDH) [1, 2].
GBMs are considered as the most common malignant brain tumors, its frequency varies between 12.0%-15.0% of all intracranial tumors, and 50.0% to 60.0% of astrocytic tumors with an annual incidence of 5.26 /100,000 population or 17,000 new diagnoses per year [3], are more common tumors in the 8th decade of life the primary type mainly, whereas the secondary affects younger ages. GBM prognosis is extremely poor as the five-year survival is only 5.0%. Patients usually survive for 12 to 15 months after the final diagnosis [4].
According to the gene expression profile GBMs can be classified into several histopathological types such as Classical, Proneural, Neural and Mesenchymal. The main gene mutations that have been recorded in GBMs are heterozygosity loss (LOH) on chromosome 10q and amplification of the epidermal growth factor receptor (EGFR). Two models have been suggested to explain the possible pathogenetic mechanisms of the tumor, the vascular occlusion model and the tumor stem cell model. The tumors that are considered to originate from glial cell include grades I, pleomorphic xanthoastrocytomas, pilocytic astrocytomas, subependyma l giant cell astrocytomas, grade II oligodendrogliomas and astrocytomas, grade III anaplastic oligodendro gliomas, anaplastic oligoastrocytomas, anaplastic astrocytomas, anaplastic ependymomas and grade IV GBM [5]. (Table 1).
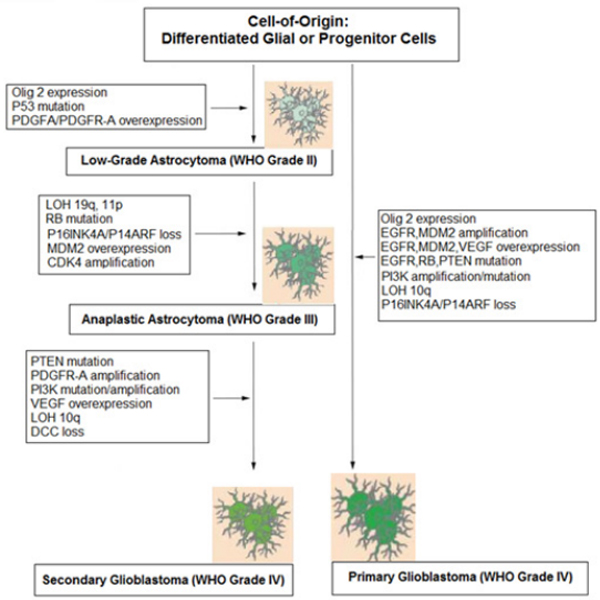
Table 1: Glioblastoma sub-types with the main genomic features.
GBM is an extremely invasive and proliferative tumor with highly abnormal vascularization, displays resistant to the common chemotherapy and radiotherapy and in general is difficult to be completely removed surgically [6].
Two molecular types have been determined, the primary GBM or de novo and the secondary that can arise due to a malignant transformation from a lower grade brain tumor and/or with the isocitrate dehydrogenase (IDH) gene mutation [7,8]. (Tables 1, 2).
Genome expression researches have been recorded several molecular and genetic abnormalities, such as Phosphatase and Tensin Homolog (PTEN) loss [9], gene amplifications at chromosome 7, deletions at chromosome 10, EGFR amplification/mutations and deletion in the locus INK4a/ ARF [10], whereas mesenchymal GBM is characterized by a very high mutation/deletion rate of Neurofibromatosis type 1(NF1), PTEN mutations, high levels of CHI3L1 and MET expression, genes involved in the Tumor Necrosis Factor (TNF) and Nuclear Factor of ?-light Polypeptide Gene Enhancer in B-cells (NFKB1) signaling pathways [11].
Proneural GBM is characterized by abnormalities, mutations mainly, of Platelet Derived Growth Factor Receptor A (PDGFRA), p53 mutations, carries the glioma CpG island methylator phenotype (GCIMP), that often is linked to isocitrate dehydrogenase (IDH)1 and 2 mutations and is characterized by common gene expression elements with secondary GBMs and
Table 2: The main molecular abnormalities in Glioblastoma according to its sub-type
.PNG)
lower grade gliomas [12]. Research of GCIMP-negative tumors showed that was contained tumors with the glioma-GCIMP phenotype that are usually IDH wild-type [13].
Proneural GBM possible has more molecular and genetic aberrants that still remain uknown [10, 11].
Isocitrate Dehydrogenase (IDH) gene mutations and GBM pathogenesis Isocitrate dehydrogenase (IDH) is a cytoplasmic enzyme in tricarboxylic acid (TCA) cycle that catalyzes isocitric acid and leads to the formation of a-ketoglutaric acid, whereas reduces coenzyme II nicotinamide adenine dinucleotide phosphate (NADPH) by oxidative decarboxylation and displays three isoforms 1,2 and 3 [14,15] .
Mutations of IDH gene result in the formation of 2-hydroxyglutaric acid from a-ketoglutaric acid. That abnormal pathway inhibits the a-ketoglutaric acid-dependent enzymes, and leads to function abnormalities of those such as epigenetic changes in a number of genes. One of those epigenetic changes is hypermethylation at a great number of genes that can lead to aberrant gene expression and inactivation of tumor suppressor's genes, molecular abnormalities that can cause GBM development [15, 16].
The most common mutations concern IDH1 isoform, whereas mutations in that enzyme consist an initiating event in gliomas pathogenesis and can separate primary from secondary GBM, however 5,0% of primary and 80,0% of secondary GBMs have such a mutation [15].
The most frequently mutation in diffuse gliomas, more than 90,0% concerns IDH1 gene and only 10,0% concerns IDH2 gene, whereas IDH3 mutations have not been found out yet [17].
The differentiation between "IDH1 mutant" and "IDH1 wild type" is important feature that is related to brain tumors, as IDH1 mutations are mainly R132H (90.0%), despite the fact that other substitutions at location R132 have also been recorded [18].
Mutations in the IDH2 gene are common finding in acute myeloid leukemia and rarely occur in glioma. In addition, mutations in IDH genes have also been found in cholangiocarcinomas and cartilaginous tumors [19].
Such mutations have been observed in 60.0%-80.0% of astrocytomas Grade II and III, oligo dendrogliomas, and oligoastrocytomas, whereas 5.0%-6.0% of primary GBMs dissemble IDH1mutations, the majority of secondary GBMs carry that mutation [15,19,20].
A better prognosis in any case of glioma grade is associated with the presence of IDH mutations [16, 17, 21].
Receptor Tyrosine Kinase (RTKs) molecular abnormalities and the specific role of Epidermal Growth Factor Receptor (EGFR) in GBM pathogenesis Receptor tyrosine kinases (RTKs) consist elements of the wide family of protein tyrosine Kinases, encompassing the receptor tyrosine kinase proteins which contain a transmembrane domain, as well as the non-receptor tyrosine kinases that do not contain transmembrane domains [22]. RTKs are the highaffinity cell surface receptors for a great number of growth factors, cytokines, and hormones. In human genome have been found out 90 unique tyrosine kinase genes, however 58 of them encode for receptor tyrosine kinase proteins [23]. They consist dominant regulators of normal cellular signaling pathways, however play a critical role in the development and progression of many types of cancer [24].
Mutations in RTKs can result to activation of a series of signaling pathways and cascades that have numerous effects on genes and proteins expression [25].
In gliomas RTKs show molecular abnormalities, such as gene amplification in a wide spectrum of RTKs, such as EGFR (60.0%- 70.0%), PDGFRA (12.0%-15.0%), and MET (5.0%) [26].
It has been mentioned previously that the EGFRvIII variant, that is present in up to 20.0% of GBM [50], is heterogeneously expressed across EGFRvIII-positive tumors [27], suggesting that its crucial contribution to gliomagenesis is inhibited by paracrine mechanisms [27,28]. It is possible that RTKs contribute to lead mitogenic cellular signaling pathways in GBM [29].
GBM cell lines after analyzing showed a co-activation of three or more RTKs. In addition, experimental trials showed variable co-expression of activated RTKs among individual cells derived from a single primary GBM. Therefore, in a single GBM exists a complicated RTK activation despite the fact that the etiology for the mentioned co-activation still remains activation despite the fact that the etiology for the mentioned co-activation still remains unknown. Researches that were focused on the structural genomics level and clonal heterogeneity in GBM cases revealed that approximately 13,0 % of GBMs that showed amplifications of EGFR, PDGFRA, or MET concealed large copy number gains in the mentioned genes [30,31].
The EGFR, known as ErbB-1, is a member of a family of closely related proteins. EGFR belongs to the family of receptors with tyrosine kinase activity (RTKs), known as HER/ErbB family and consists of 4 known receptors: EGFR or HER1-4/ErbB1-4. The EGFR gene, located on chromosome 7p12, encodes for a transmembrane tyrosine kinase receptor. Its activation by appropriate ligand results in the activation of the receptor's intracellular tyrosine kinase domain which undergoes autophosphorylation and is able to trigger a cascade of intracellular events that promote cell proliferation and migration [32].
EGFR gene amplification has been observed in 40,0-50,0% of the classical GBM type [33] and 63,0-75,0% of GBM cases transfer EGFR gene rearrangements that lead to tumors characterized by wild-type EGFR and mutated EGFR [34].
A splice variant that is responsible for a EGFR mutant form has been observed in 20,0-50,0% of GMB-induced by EGFR gene amplification , whereas in anaplastic astrocytomas, the incidence of EGFR amplification is 17,0% . The presence of EGFR gene amplification in GBM cases is associated with poor clinical prognosis [35]. However, in contrast to EGFR mutants in lung cancer, treatment with EGFR tyrosine kinase inhibitors (TKIs) appears to be less successful in GBM compared with lung cancer [36, 37].
Increased expression of other receptor tyrosine kinases (RTK) such as PDGFRA and Vascular Endothelial Growth Factor (VEGFR) contribute to growth of GBM through the activation of RAS/ERK or phosphatidylinositide-3 kinase (PI3K)/AKT-dependent signaling pathways [35].
Cells that carry EGFR amplification usually show different types of EGFR mutations, such as EGFRvIII rearrangement, that is due to deletion of exons 2-7 in EGFR mRNA and leads to an in-frame deletion of 267 amino acids in the extracellular domain of EGFR and it results to be unable to bind ligand [38]. EGFRvIII rearrangement decreases kinase degradation and over-activates the downstream targets of signaling pathways. In GBM patients, EGFRvIII mutation indicates poor prognosis [39].
Recent investigations have shown that EGFR wild type is necessary for the oncogenic effect of EGFRvIII. Dimerization of EGFRvIII is required for its activation and EGFRvIII may homoor heterodimerize with EGFR. As a matter of fact exists a feedforward loop as EGFRvIII is activated in case wild type EGFR is co-expressed. Heparin-binding epidermal growth factor (HBEGF) activates EGFR wild type and this activation induced by EGFRvIII [40].
EGFRvIII increased activation results to create an increased transactivation of several RTK families such as EphA2 and Met that may have possible involvement in EGFRvIII oncogenesis [41]. Recently, it has been found out in individual GBM cells an EGFR-EGFRvIII-STAT3 signaling axis that co-amplify EGFR and EGFRvIII. It has also been shown that exists a co-operation between EGFR and EGFRvIII in case of in vivo transformation and EGF treatment of cells that express EGFR and EGFRvIII could lead to phosphorylation of both kinases. It is clear that EGFR promotes in a single direction EGFRvIII signaling in GBM cells. EGFR-catalyzed phosphorylation of EGFRvIII has as a result its transportation into the nucleus and an increased production of a complex between EGFRvIII and STAT3. The last observation suggest that the mentioned kinases increase and continue for a long time STAT3 activity in the nucleus.Therefore is possible that increased EGFR levels even in the absence of EGFRvIII could unsettle that function, whereas targeting EGFR in combination with STAT3 signaling may be a treatment direction for EGFRvIII-positive GBM patients [42].
It has been shown the presence of multiple EGFR variants in a single GBM tumor focusing on the intratumoral heterogeneity of GBM that could be attributed to the plasticity of EGFR amplicons [43].
EGFRvII is produced by deletion of exons 2-7 of the EGFR gene and can be appeared in 9.0% of GBM cases that are focally amplified. EGFRvII's constitutive expression can lead to AKT downstream activation signaling, according to that of EGFRvIII, but without increased activation of ERK. There has been a possibility EGFRvII may activate an alternative signaling pathway such as STAT3 and set a more direct way to induce alterations in transcription. It seems that EGFRvII is able to increase EGFR Tyrosine Kinase Inhibitors' (TKIs) sensitivity [43, 44].
In 90.0% of GBMs cases PI3K molecular signaling pathway is hyperactivated due to molecular aberrants such as activating mutation or EGFR amplification and PTEN gene loss [45]. PI3K signaling pathway regulates anti-apoptotic mechanisms and promotes tumor growth and survival, including through sterol regulatory element binding protein 1 (SREBP-1)-dependent lipogenesis [46].
Promoter methylation of MGMT (O6-methyl guanine-DNA methyltransferase) and GBM O6- methyl guanine-DNA methyltransferase (MGMT) is a protein that is encoded by the MGMT gene that is located on chromosome 10q26 and is crucial for genome stability. It repairs the naturally occurring mutagenic DNA damages and prevents mismatch and errors during DNA replication and transcription. Consequently, loss of MGMT increases the oncogenesis risk after exposure to alkylating agents. MGMT is transferred into the nucleus in case of DNA damage, it binds to O6-methyl-guanine and demethylates the alkylating agent after interaction with it [47].
Epigenetic events, as genes methylation can lead to inactivation of tumor suppressor genes, DNA repair genes, and proapoptotic genes, abnormalities that can induce carcinogenesis [48, 49].Approximately 50.0 % of MGMT promoter methylation have been observed in the secondary GBMs, and mainly in elder patients [50].
MGMT promoter methylation status is considered as a survival predictive biomarker especially in elderly GBM patients, whereas in younger ones the pro-neural GBM type that characterized by hypermethylation of CpG sites is associated with better prognosis, as is well known that epigenetic mechanisms also have prognostic significance in GBM patients [37].
Telomerase Reverse Transcriptase Gene (hTERT) genetic mutations and GBM Telomerase consists a ribonucleoprotein enzyme complex that maintains and prolongs chromosome ends- telomeres of the eukaryotic chromosomes using one native RNA molecule as a template and thus is able to extend the number of cell divisions [51]. It acts as a RNA dependent-DNA- polymerase that can compensate for the loss of those DNA sequences by producing telomeric replicates in cells that still have the ability to be divided. A catalytic subunit,telomerase reverse transcriptase (hTERT) with the telomerase RNA component (TERC) comprises the most important unit of the telomerase complex [52].
Mutations in the hTERT have been observed in about 75.0% of GBM cases. Despite the fact that hTERT mutation status itself cannot be a predictive or prognostic factor for GBM treatment results, it is possible that the positive prognosis attributed to MGMT promoter methylation in GBM patients may depend on simultaneous hTERT promoter mutation [53, 54].
RB1 mutation and deletion in GBM The retinoblastoma protein pRb encoded by RB1 gene that is located on chromosome 13, consists a tumor suppressor protein that is associated with abnormalities in several types of cancers. Its main function is to prevent excessive cell growth by inhibiting cell cycle progression until a cell is ready to divide. At that point of the cell cycle, pRb is phosphorylated, becomes inactive and allows cell cycle progression as binding and repressing E2F transcriptional targets. It is also a recruiter of several chromatin remodeling enzymes such as methylases and acetylases. The CDKN2A/p16-CDK4/6-RB pathway characterized by molecular abnormalities in GBM patients [55]. Those abnormalities play a critical role in gliomagenesis or umor progression from lower-grade astrocytomas [56]. CDK4 and CDK6 phosphorylate RB1, that induces the release of the transcription factor E2F, thus facilitating the transition of the cell cycle from the G1 to S phase and this process is negatively regulated by the p16 protein which binds and inhibits CDK4/6 unction.Consequently, expression abnormalities of p16, CDK4, CDK6, or RB1 can lead to cell cycle dysregulation [57]. It has been shown that this pathway is altered in 80% of primary GBMs and the most common genetic abnormality concerns CDKN2A gene mutation or deletion, CDK4 amplification, and RB1 mutation or deletion. Rb promoter methylation is another epigenetic alteration that has been shown in 14,0% of primary GBM and 43,0% of secondary ones [58].
Genetic Mutations of Phosphatase and Tensin Homolog (PTEN) and GBM PTEN consists a protein that, is encoded by the PTEN gene located on chromosome 10q23 and acts as a tumor suppressor gene through the action of its phosphatase protein product, that is involved in the cell cycle regulation, preventing cells proliferation and division. Mutations of this gene can lead in the development of several types of cancer [59, 60].
The protein encoded by PTEN gene is a phosphatidylinositol-3, 4, 5-trisphosphate 3-phosphatase (PIP3). It contains a tensin-like domain and a catalytic domain similar to that of the dual specificity protein tyrosine phosphatases and dephosphorylates phosphoinositide substrates. Thus, negatively regulates intracellular levels of phosphatidylinositol-3, 4, 5-trisphosphate in cells and acts as a tumor suppressor by negatively regulating Akt/PKB signaling pathway. PTEN is also able to inhibit cell invasion, cell adherence to the surrounding matrix and vascularization, whereas in case of excessive cell differentiation, it has the ability to regulate cell cycle and induce apoptosis. Consequently contributes to inhibition of tumor growth [60].
Approximately 86.0% of GBM patients have lost PTEN gene expression and show molecular abnormalities in receptor tyrosine kinase/phosphoinositide 3-kinase (RTK/PI3K) signaling pathway that it regulates cell survival, cell proliferation, adhesion, cell mobility and differentiation. Patients that suffered from primary GBM display PTEN point mutation in 26,0%- 34,0% ,whereas in anaplastic astrocytomas patients the PTEN mutation rate is approximately 18,0%. It is important to mention that GBM patients with PTEN mutation have poor prognosis [61].
Mutation and deletion of Neurofibromatosis type 1 (NF1) gene and GBM NF1 gene is located on chromosome 17q11.2 and encodes for the neurofibromatosis protein, acts as a potential tumor suppressor , regulates negatively the Ras oncogene and consists a mechanistic target of rapamycin (mTOR) signaling pathways in astrocytes. In GBM patients is observed inactivation of that gene (type 1) due to excessive degradation by the ubiquitinproteasom pathway, whereas it's genetic loss consists the second reason that is able to lead to excessive cell proliferation and oncogenesis [62].
NF1 loss leads to increased activity in crucial pro-tumorigenic signaling pathways, particularly the Mitogen-Activated Protein Kinase (MAPK) pathway. NF1 patients are predisposed to several types of tumors affecting the central and peripheral nervous system such as pilocytic astrocytoma whereas adults patients, may develop gliomas of all types and grades [63]. A retrospective study of NF1-associated gliomas showed that 27.0% were diffusely infiltrating astrocytomas, and 22.0% were high grade gliomas, such as anaplastic astrocytomas and GBMs [64]. Another study that was based on mice was found that NF1 and Tp53 co-inactivation led to GBM development and was characterized by high penetration [65, 66].
Mutations are more common in mesenchymal type of GBM [67], whereas experimental trials in mice showed that NF1 deletion could lead to an excessive astrocytes proliferation and migration as a result of mTOR transactivation that is mediated by the Ras oncogenic signaling pathway. Mutations or deletions of that gene, lead to regulation of STAT3 signaling pathway, that consist potential down-stream target genes, by the mechanistic Target Of Rapamycin Complex 1(mTORC1) and Ras-related C3 botulinum toxin substrate 1 (Rac1) and enhance cyclin D1 levels that are responsible for an excessive cell proliferation [68].
In astrocytes of genetically engineered mice models double hit of NF1gene can lead in vivo and in vitro to cell proliferation, however it is not adequate to induce GBM [69], whereas in case of homozygous NF1 deletions (NF1-/-), astrocytes need heterogeneity to induce GBMs [70].
NF-kB signalling in GBM The nuclear factor NF-kB is a transcription factor that is activated by the EGFR signaling pathway and is involved in the evasion of apoptosis (programmed cell death). It is also activated by inflammatory cytokines (TNF-α, IL-1β, IFN-γ) and bacterial products (LPS), consists an important effector of the inflammatory response as regulates normal immune response, shows anti-apoptotic action by interaction with Bcl-2 and Bcl-XI factors and is associated with chronic nflammation and carcinogenesis of the respiratory tract, etc. and maybe the key that allows preneoplasmatic and malignant cells to escape from apoptosis [71].
Its abnormal constitutive activation has been observed in GBM [72], whereas the encoding nuclear factor of k-light polypeptide gene enhancer in B-cells inhibitor-A (NKFBIA) is a NF-kB inhibitor that abolish signaling in the NF-?B and EGFR pathways and plays a crucial role as a suppressor in GBM cases. Deletion of the nuclear factor NFKBIA has an effect that is similar to amplification of EGFR in the pathogenesis of GBM. A two-gene model that was based on NFKBIA expression and MGMT expression was strongly associated with the clinical course of the disease [73].
The role of Programmed Cell Death Protein 4 (PDCD4) protein in GBM Programmed Cell Death Protein 4 (PDCD4) is a nuclear/cytoplasmic protein that encoded by the PDCD4 gene that consists a tumor suppressor gene, inhibits translation by interacting with translation initiation factor eIF4A and inhibits its RNA helicase activity that has been involved in the development of several types of cancer. In the nucleus, PDCD4 affects the transcription of specific genes by modulating the activity of several transcription factors [74, 75].
PDCD4 interacts with nuclear factor NF-kB in GBM cases. To be more specific, PDCD4 interacts with p65 and inhibits NFkB nuclear localization and consequently, inhibits NF-?B transcriptional activation [76].
PDCD4 protein regulates Matrix Metalloproteinase-9 (MMP-9) and VEGF that are NF-kB target genes and are overexpressed in GBM tissues [76,77].
Astrocyte Elevated Gene-1 protein (AEG-1) expression and GBM Astrocyte elevated gene-1 protein (AEG-1) is a protein that is encoded by the MTD gene [78]. AEG-1 acts as an oncogene in several malignant tumors such as elanoma, malignant glioma, breast cancer and hepatocellular carcinoma. It is highly expressed in those cancers contributes to their development and progression, induced by c -Myc oncogene and plays a crucial role in independent proliferation of cancer cells [79]. AEG-1 plays a critical role in tumorigenesis in several organs as already has reported, and consists a Ras target and activates many oncogenic cellular signaling pathways such as MAPK/ ERK, PI3K-AKT, NF?B and Wnt, that regulate cellular functions including invasion, cell proliferation, angiogenesis, metastasis and chemoresistance [80,81].
AEG-1 was firstly identified as an human immunodeficiency virus (HIV-1) and tumor necrosis factor A(TNF-A)-inducible transcript in primary human fetal astrocytes and showed a significant interest in the field of cancer as its expression was found to be increased in subsets of breast cancer, GBM and melanoma cells, whereas plays a central role in Ha-ras-mediated oncogenesis through PI3K/Akt signaling pathway, promotes the transformation of immortalized melanocytes and is overexpressed in more than 90,0 % of brain tumors [82]. In addition, AEG-1 also ctivates the VEGF promoter in malignant glioma cells [83].
As already has been reported AEG-1 expression is increased in several types of cancers, whereas its overexpression protects primary and transformed human and mice cells from serum starvation-induced apoptosis due the activation of PI3K/ Akt signaling pathway. Those observations suggest, that AEG1 may function as an oncogene [83].
Its expression was also elevated in adult astrocytes transformed by SV40-T antigen, hTERT, and oncogenic Ha-ras that displayed an aggressive glioma-like phenotype [84].
Regarding the relationship between AEG-1 and the Excitatory Amino Acid Transporter 2 (EAAT2) that consists one of the major glutamate transporters expressed predominantly in astroglial cells, a strong negative association between expression of AEG-1 and the EAAT2 has been reported . EAAT2 suppression is responsible for a reduction of uptake of glutamate by glial cells, and leads to induction of necrosis of neuronal cell that contributes to glioma-induced neurodegeneration. In addition, AEG-1 promotes gliomagenesis, particularly based on the main characteristics of glioma that are tumor growth and invasion [85].
AXL receptor tyrosine kinase overexpression and GBM Tyrosine-protein kinase receptor is an enzyme that in humans is encoded by the AXL gene [86]. It has been proposed that AXL facilitates immune escape and drug-resistance by cancer cells, functions that can lead to aggressive and metastatic cancers [87].
It normal function is associated with signals transduction from the extracellular matrix into the cytoplasm by binding growth factors like vitamin K-dependent protein growth-arrest-specific gene 6 (GAS6), whereas is also involved in the stimulation of cell proliferation and survival [88] as activates downstream signaling pathways that include PI3K-AKT-mTOR, MEK/ERK, F-kB, and JAK/STAT [89].
AXL overexpression in astrocytoma/glioblastoma has been reported according to recent studies [90,91] and especially its overexpression in astrocytoma cell invasion and migration [91], however the functional influences of AXL expression in astrocytoma that are associated with signaling, cell survival, and cell proliferation still remain unclear. In addition, it has not been confirmed a role for the Mer RTK in astrocytoma, whereas it has been recorded an abnormal expression of Mer and AXL RTKs in astrocytoma cell lines and primary patient samples, and the role of AXL in cell survival, proliferation, and migration. Mer and AXL receptor tyrosine kinases are expressed at abnormally elevated levels in a wide spectrum of cancers, and those receptors are able to activate crucial antiapoptotic signaling pathways that can promote oncogenesis. Inhibition of either Mer or AXL RTK expression can lead to extremely different phenotype compared with the parental astrocytoma cells, with enhanced apoptosis and apoptotic signaling, decreased cell proliferation and growth, and higher sensitivity to chemotherapeutic agents [92].
TP53 promoter methylation and mutations in GBM The TP53 gene encodes for a protein that is involved in the cell cycle, response to DNA damage, cell differentiation, and cell death, and is responsible for genomic stability [93]. Mutations of the TP53 gene often cause changes in the functions of the protein and lead to its inactivation and accumulation of mutated protein in the nuclei of tumor cells [94].
The TP53 mutation have been observed in about 30.0% of cases of primary GBM and in 65.0%-90,0% of cases of secondary GB, and especially 59,0% in low-grade astrocytoma and 53,0% in anaplastic astrocytoma, finding that indices that TP53 mutation occurs early in tumorigenesis [95].
TP53 point mutations are equally concern the 5-8 exons (hotspot mutations) in cases of primary GBM, whereas in secondary GB most frequent point mutation are distributed at codon 248 and 273 (exon 7 and 8), and in CpG sites (methylation region) [96].
The abnormal function of the mutated TP53 can lead not only in mutations, but can causes abnormalities in cellular signaling pathway s that are involved genes such as MDM2, MDM4, or CDKN2A/p14ARF, that binds to MDM2 and inhibits MDM2- mediated TP53 degradation. It has been recorded that in 87.0% of GBMs were recorded molecular abnormalities that concern the TP53/MDM2/CDKN2A signaling pathway [58].
Proneural type of GBM that characterized by high expression of oligodendrocytic genes was carried the majority of TP53 mutations and TP53 heterozygosity loss 54.0% [97]. In primary GBM, has been found an association between EGFR amplification and absence of TP53 mutation as is considered mutually exclusive [96].
MGMT promoter methylation is associated with GBM pathogenesis, as already has been reported, however that alteration was linked to the presence of G: C to A: T transition mutations in TP53 in many types of cancer, including GBM [98].
Tripartite Motif (TRIM) proteins and GBM Tripartite motif (TRIM) proteins are members of the E3 ubiquitin ligases family that contain a TRIM encompassing RING finger domain, one or two zinc-binding B-box domains and coiled coil domains and are encoded by the TRIM3 gene that is located on chromosome 11p15.5 [99]. TRIM proteins are responsible for cellular functions as regulate cell proliferation, apoptosis and transcriptional regulation [100].
TRIM3 was firstly identified and determined as a brain-enriched RING finger protein [99]. 25% of GBMs and lowergrade gliomas showed TRIM3 loss caused by its deletion or DNA methylation. TRIM3 deletion is highly associated with the GBM proneural transcriptional type that is enriched for genes that regulate neural development and proliferation. In the same investigation was suggested that TRIM3 regulates cell proliferation and differentiation through the Musashi/Numb/ Notch/Hedgehog signaling pathway, as well as c-Myc, and its loss increases the glioma stem-cell population by disrupting asymmetric cell division and cellular differentiation [101].
Loss of Heterozygosity (LOH) as additional genetic alteration that contributes to GBM pathogenesis LOH on chromosome 10 consists the most frequent genetic alteration in GBMs, and occurs in 80% of cases, however rarely occurs in low-grade astrocytomas and it accounts for 40% of the anaplastic astrocytomas. It has been shown that the majority of GBMs have lost an entire copy of chromosome 10. Partial LOH GBMs show three common deletion on10p14-pter [102], 10q23-24 [103] and 10q25-qter [102].
LOH 10 has been observed in 60%-100% of GBMs with EGFR amplification [104] and in 40%- 80% of GBMs with a p53 mutation [104] and this finding leads to the suggestion that LOH 10 is implicated in primary and secondary GBMs' pathogenesis, whereas secondary GBMs with LOH 10 do not have loss of chromosome 10p but are characterized by partial or complete loss of chromosome 10q.
LOH on 22q is a common alteration in 11-39% of gliomas, uncommon in low-grade astrocytomas, but is more frequently found in anaplastic astrocytomas and GBMs. That alteration occurs more frequently in the advanced stage of several types cancers and it suggests a link between 22q LOH and tumor progression [105].
LOH on chromosome 19q has been recorded in 70% of oligodendrogliomas, and in 50% of oligoastrocytomas, in15% of diffuse low-grade astrocytomas, in nearly 45% of anaplastic astrocytomas, and 25% of unselected GBMs , finding that could be attributed to the fact that a significant percentage of unselected GBMs does not develop through LOH 19q [106,107].
It was also found that approximately 29% of primary GBMs, tumors from an initial resection and 40% of recurrent GBMs, tumors from a non-initial surgical resection characterized by LOH on chromosome19q.That alteration is frequently implicated in the progression from low-grade astrocytomas to secondary GBMs, but is not typically involved in the development of primary GBMs. Even though no evidence exists, the higher frequency of LOH 19q in the previous study could be suggested that in the pathway that leads to secondary GBMs, LOH 19q consists an alteration that takes place after LOH on chromosome 10 [107]. LOH on chromosome 10q25-qter is associated with obtainment of GBM phenotype, but LOH on chromosome 19 was observed in only 1 of 5 cases with foci showing a sudden histological transition from low-grade or anaplastic astrocytoma to GBM [107].
LOH on chromosome 1p has been detected in nearly 10% of low-grade astrocytomas, in 20% of anaplastic astrocytomas, and 10% of GBMs [107-109]. On that chromosome have been detected genes that are involved in cancer pathogenesis, such as p73 [110] at 1p36.33, E2F2 [111], TR2 [112], TNFR2 [113], DR3 [114], and DR 5 [115] at 1p36, and RAD54 [116] and p18 (INK4C) [117] at 1p32. Loss of the 1p36 locus has been recorded in oligodendrogliomas (70%) and oligoastrocytomas (50%) [107,118].
Oligodendrogliomas with LOH on 1p often show concurrent LOH on 19q [118,119], finding that leads to a suggestion of a cooperation between those genetic alterations. In the previous study on GBMs, there was no significant association between LOH on chromosomes 1p and 19 [118]. LOH on chromosome13q has been recorded in almost 20% of lowgrade astrocytomas [55,120], 25% of anaplastic astrocytomas [55,120], and 35% of GBMs [55,120,121].The Retinoblastoma gene (RB) is located at 13q14.2 [122] and the breast cancer susceptibility locus 2 (BRCA2 ) on 13q12.1 [123,124]. Only in 10% of GBMs [55, 120,125] have been recorded mutations that concern the RB gene, finding that suggests the presence of other tumor suppressor gene on this chromosome.
REFERENCES
- Kleihues P, Burger PC, Aldape KD, Brat DJ, et al. (2007). Glioblastoma in: WHO classification of tumours of the central nervous system (4th edn). International Agency for Research on Cancer (IACC) Lyon.
- Vauleon E, Avril T, Collet B, Mosser J, et al. (2010). Overview of cellular immunotherapy for patients with glioblastoma. Clin Dev Immunol. 2010.
- Omuro A and De Angelis LM. (2013). Glioblastoma and other malignant gliomas: a clinical review. J Am Med Assoc. 310(17):1842-1850.
- Ohba S and Hirose Y. (2016). Current and Future Drug Treatments for Glioblastomas. Curr Med Chem. 23(38): 4309-4316.
- Schwartzbaum JA, Fisher JL, Aldape KD and Wrensch M. (2006). Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2(9): 494-503. quiz 491 p following 516.
- Weller M, van den Bent M, Hopkins K, Tonn JC, et al. (2014). EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 15(9): e395-e403.
- Veliz I, Loo Y, Castillo O, Karachaliou N, et al. (2015). Advances and challenges in the molecular biology and treatment of glioblastoma-is there any hope for the future? Ann Transl Med. 3(1): 7.
- Scherer HJ. (1940). A Critical Review: The Pathology of Cerebral Gliomas. J Neurol Psychiatry. 3(2): 147-177.
- Rosell R, de Las Penas R, Balana C, Santarpia M, et al. (2008).Translational research in glioblastoma multiforme: molecular criteria for patient selection. Future Oncol. 4(2): 219-228.
- Theeler BJ, Yung WK, Fuller GN and De Groot JF. (2012). Moving toward molecular classification of diffuse gliomas in adults. Neurology. 79(18): 1917-1926.
- Bredel M, Scholtens DM, Harsh GR, Bredel C, e t al. (2009). A network model of a cooperative genetic landscape in brain tumors. J Am Med Assoc. 302(3). 261-275.
- Verhaak RG, Hoadley KA, Purdom E, Wang V, et al. (2010). Integrated genomic analysis identifes clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 17(1): 98- 110.
- Ozawa T, Riester M, Cheng YK, Huse JT, et al. (2014). Most Human Non-GCIMP Glioblastoma Subtypes Evolve from a Common Proneural-like Precurso Glioma. Cancer Cell. 26(2): 288-300.
- Ichimura K. (2012). Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathology. 29(3): 131-139.
- Hartmann C, Hentschel B, Wick W, Capper D, et al. (2010). Patients with IDH wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 120(6): 707-718.
- Chen R, Ravindra VM, Cohen AL, Jensen RL, et al. (2015). Molecular features assisting in diagnosis, surgery, and treatment decision making in low-grade gliomas. Neuro surg Focus. 38(3): E2.
- Ohgaki H and Kleihues P. (2013). The definition of primary and secondary glioblastoma. Clin Cancer Res. 19 (4): 764-772.
- Yan H, Parsons DW, Jin G, McLendon R, et al. (2009). IDH1 and IDH2 mutations in gliomas. N Engl J Med. 360: 765- 773.
- Dunn GP, Rinne ML, Wykosky J, Genovese G, et al. (2012). Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 26(8): 756-784.
- Hartmann C, Meyer J, Balss J, Capper D, et al. (2009). Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 118(4): 469-474.
- Szopa W, Burley TA, Kramer-Marek G and Kaspera W. (2017). Diagnostic and Therapeutic Biomarkers in Glioblastoma: Current Status and Future Perspectives. Biomed Res Int. 2017. 8013575.
- Hubbard SR and Till JH. (2000). Protein tyrosine kinase structure and function. Ann Rev Biochemistry. 69: 373-398.
- Robinson DR, Wu YM and Lin SF. (2000). The protein tyrosine kinase family of the human genome. Oncogene. 19 (49): 5548-5557.
- Zwick E, Bange J and Ullrich A. (2001). Receptor tyrosine kinase signaling as a target for cancer intervention strategies". Endoc Rel Cancer. 8 (3): 161-173.
- Lemmon MA and Schlessinger J. (2010). Cell signaling by receptor tyrosine kinases. Cell. 141 (7): 1117-1134.
- Brennan CW, Verhaak RG, McKenna A, Campos B, et al. (2013). The Somaticgenomic landscape of glioblastoma. Cell. 155(2): 462-477.
- Nishikawa R, Sugiyama T, Narita Y, Furnari F, et al. (2004). Immunohistohistochemical analysis of the mutant epidermal growth factor, delta EGFR, in glioblastoma. Brain Tumor Pathol. 21(2): 53-56.
- Inda MM, Bonavia R, Mukasa A, Narita Y, et al. (2010). Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 24(16): 1731-1745.
- Stommel JM, Kimmelman AC, Ying H, Nabioullin R, et al. (2007). Co activation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 318(5848): 287-290.
- Snuderl M, Fazlollahi L, Le LP, Nitta M, et al. (2011). Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 20(6): 810-817.
- Szerlip NJ, Pedraza A, Chakravarty D, Azim M, et al. (2012). Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplifcation in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci USA. 109(8): 3041-3046.
- Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, et al. (2007). The epidermal growth factor receptor: from development to tumorigenesis. Differentiation. 75(9): 770-787.
- Kohsaka S, Wang L, Yachi K, Mahabir R, et al. (2012). STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol Cancer Ther. 11(6): 1289-1299.
- Gan HK, Cvrljevic AN and Johns TG. (2013). the epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBSJ. 280(21): 5350-5370.
- Aldape K, Zadeh G, Mansouri S, Reifenberger G, et al. (2015). Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 129(6): 829-848.
- Barkovich KJ, Hariono S, Garske AL, Zhang J, et al. (2012). Kinetics of inhibitor cycling underlie therapeutic disparities between EGFR-driven lung and brain cancers. Cancer Discov. 2(5): 450-457.
- Vivanco I, Robins HI, Rohle D, Campos C, et al. (2012). Differential sensitivity of glioma- versus lung cancer-specifc EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2(5): 458-471.
- Hatanpaa KJ, Burma S, Zhao D and Habib AA. (2010). epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 12(9): 675- 684.
- Atsumi S, Nosaka C, Adachi, Kimura T, et al. (2016). New anti-cancer chemicals Ertredin and its derivatives, regulate oxidative phosphorylation and glycolysis and suppress sphere formation in vitro and tumor growth in EGFRvIII-transformed cells. BMC Cancer. 16: 496.
- Li L, Chakraborty S, Yang CR, Hatanpaa KJ, Cipher DJ, et al. (2014). An EGFR wild type EGFRvIII-HB-EGF feed-forward loop regulates the activation of EGFRvIII. Oncogene. 33(33): 4253-4264.
- Johns TG, Perera RM, Vernes SC, Vitali AA, et al. (2007). the efficacy of epidermal growth factor receptor-specifc antibodies against glioma xenografts is influenced by receptor levels, activation status, and heterodimerization. Clin Cancer Res. 13(6): 1911-1925.
- Fan QW, Cheng CK, Gustafson WC, Charron E, et al. (2013). EGFR phosphorrylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell. 24(4): 438-449.
- Francis JM, Zhang CZ, Maire CL, Jung J, et al. (2014). EGFR Variant Hetero geneity in Glioblastoma Resolved through Single Nucleus Sequencing. Cancer Discov.4 (8): 956-971.
- Gini B, Mischel PS. (2014). Greater Than the Sum of Its Parts: Single-Nucleus Sequencing Identifes Convergent Evolution of Independent EGFR Mutants in GBM. Cancer Discov. 4(8): 876-878.
- Chakravarti A, Zhai G, Suzuki Y, Sarkesh S, et al. (2004). The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J Clin Oncol. 22(10): 1926-1933.
- Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, et al. (2011). An LXR Agonist Pro motes Glioblastoma Cell Death through Inhibition of an EGFR/AKT/SREBP-1/LDLRDependent Pathway. Cancer Discov. 1(5): 442-456
- Dullea A and Marignol L. (2016) .MGMT testing allows for personalized therapy in the temozolomide era. Tumour Biol. 37(1): 87-96.
- Cankovic M, Nikiforova MN, Snuderl M, Adesina AM, et al. (2013). The role of MGMT testing in clinical practice: a report of the association for molecular pathology. J Mol Diagn. 15(5): 539-555.
- Eoli M, Menghi F, Bruzzone MG, De Simone T, et al. (2007). Methylation of O6- Methylguanine DNA Methyltransferase and Loss of Heterozygosity on 19q and/or 17p are Overlapping Features of Secondary Glioblastomas with Prolonged Survival. Clin Cancer Res. 13(9): 2606-2613
- Jordan JT, Gerstner ER, Batchelor TT, Cahill DP, et al. (2016). Glioblastoma care in the elderly. Cancer. 122(2): 189-197.
- Yao Y, Choi J and Parker I. (1995). Quan tal puffs of intracellular Ca2+ evoked by inositol trisphosphate in Xenopus oocytes. J Physiol 482 (pt 3): 533-553.
- Hiyama K, Hiyama E, Ishioka S, Yamakido M, et al. (1995) Telomerase activity in small-cell and non-small-cell lung cancers. J Natl Cancer Inst. 87(12): 895-902.
- Arita H, Yamasaki K, Matsushita Y, Nakamura T, et al. (2016). A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroupsof newly diagnosed glioblastomas. Acta Neuropathol Commun. 4(1): 79.
- Nguyen HN, Lie A, Li T, Chowdhury R, et al. (2017). Human TERT promoter mutation enables survival advantage from MGMT promoter methylation in IDH1 wild-type primary glioblastoma treated by standard chemo radiotherapy. Neuro Oncol. 19(3): 394-404.
- Ichimura K, Schmidt EE, Goike HM and Collins VP. (1996). Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene. 13(5): 1065-1072.
- Chow LM, Endersby R, Zhu X, Rankin S, et al. (2011). Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell. 19(3): 305-316.
- Biernat W, Tohma Y, Yonekawa Y, Kleihues P, et al. (1997). Alterations of cell cycle regulatory genes in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 94(4): 303-309.
- The Cancer Genome Atlas Network. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 455(7216): 1061- 1068.
- Steck PA, Pershouse MA, Jasser SA, Yung WK, et al. (1997). Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers". Nature Genetics. 15 (4): 356-362.
- Chu EC and Tarnawski AS (2004). PTEN regulatory functions in tumor suppression and cell biology. Med Sci Monitor. 10 (10): RA235-RA241.
- Han F, Hu R, Yang H, Liu J, et al. (2016). PTEN gene mutations correlate to poor prognosis in glioma patients: a meta-analysis. Onco Targets Ther. 9: 3485-3492.
- McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, et al. (2009). Protea somal and Genetic Inactivation of the NF1 Tumor Suppressor in Gliomagenesis. Cancer Cell. 16(1): 44-54.
- Gutmann DH, James CD, Poyhonen M, Louis DN, et al. (2003). Molecular analysis of astrocytomas presenting after age 10 in individuals with NF1. Neurology. 61(10): 1397-1400.
- Rodriguez FJ, Perry A, Gutmann DH, O'Neill BP, et al. (2008). Gliomas in neuro-fibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol.67 (3): 240-249.
- Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, et al. (2000). Nf1; Trp53 mutantmice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 26(1):109- 113.
- Zhu Y, Guignard F, Zhao D, Liu L, et al. (2005). Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 8(2): 119-130.
- Banerjee S, Byrd JN, Gianino SM, Harpstrite SE, et al. (2010). The Neuro fibromatosis Type 1 Tumor Suppressor Controls Cell Growth by Regulating Signal Transducer and Activator of Transcription-3 Activity In vitro and In vivo. Cancer Res.70 (4): 1356-1366.
- Bajenaru M, Zhu Y, Hedrick N, Donahoe J, et al. (2002). Astrocyte-Specific Inactivation of the Neurofibromatosis 1 Gene (NF1) Is Insufficient for Astrocytoma Formation. Mol Cell Biol. 22(14): 5100-5113.
- Solga AC, Gianino SM and Gutmann DH. (2014). NG2-cells are not the cell of origin for murine neurofibromatosis-1 (Nf1) optic glioma. Oncogene. 33(3): 289-299.
- Vizcaino MA, Shah S, Eberhart CG and Rodriguez FJ. (2015). Clinicopathologic implications of NF1 gene alterations in diffuse gliomas. Hum Pathol. 46(9): 1323-1330
- Stathopoulos GT, Sherrill TP, Cheng DS, Scoggins RM, et al. (2007). Epithelial NF kappaB activation promotes urethane-induced lung carcinogenesis. Proc Natl Acad Sci USA. 104(47): 18514-18519.
- Bredel M, Bredel C, Juric D, Duran GE, et al. (2006). Tumor necrosis factor alpha induced protein 3 as a putative regulator of nuclear factor-kappaB-mediated resistance to O6-alkylating agents in human glioblastomas. J Clin Oncol. 24(2): 274-287.
- Bredel M, Scholtens DM, Yadav AK, Alvarez AA, et al. (2011). NFKBIA deletion in glioblastomas. N Engl J Med. 364: 627-637.
- Soejima H, Miyoshi O, Yoshinaga H, Masaki Z, et al. (1999). Assignment of the programmed cell death 4 gene (PDCD4) to human chromosome band 10q24 by in situ hybrid dization. Cytogen Gen Res. 87(1-2): 113-114.
- Yang HS, Cho MH, Zakowicz H, Hegamyer G, et al. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol. 24(9): 3894-3906.
- Hwang SK, Baker AR, Young MR and Colburn NH. (2014). Tumor suppressor PDCD4 inhibits NF-kappaB-dependent transcription in human glioblastoma cells by direct interaction with p65. Carcinogenesis. 35(7): 1469-1480.
- Zanotto-Filho A, Braganhol E, Schroder R, de Souza LH, et al.(2011). NFKap-paB inhibitors induce cell death in glioblastomas. Biochem Pharmacol. 81(3): 412-424.
- Brown DM and Ruoslahti E. (2004). Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell. 5(4): 365-374.
- Yoo BK, Emdad L, Su ZZ, Villanueva A, et al. (2009). Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. Journal Clin Invest. 119(3): 465-477.
- Sarkar D, Emdad L, Lee SG, Yoo BK, et al. (2009). Astrocyte elevated gene-1: far more than just a gene regulated in astrocytes. Cancer Res. 69(22): 8529-8535.
- Lee SG, Su ZZ, Emdad L and Sarkar D, et al. (2006). Astrocyte elevated gene-1 (AEG-1) is a target gene of oncogenic Ha-ras requiring phosphatidylinositol 3-kinase and c-Myc. Proc Natl AcadSci USA. 103(46): 17390-17395.
- Emdad L, Sarkar D, Su ZZ, Randolph A, et al. (2006). Activation of the Nuclear FactorKB Pathway by Astrocyte Elevated Gene-1: Implications for Tumor Progression and Metastasis. Fisher Cancer Res. 66(3): 1509-1516.
- Emdad L, Lee SG, Su ZZ, Jeon HY, et al. (2009). Astrocyte elevated gene-1(AEG-1) functions as an oncogene and regulates angiogenesis. Proc Natl Acad Sci USA. 106(50): 21300-21305.
- Kang DC, Su ZZ, Sarkar D, Emdad L, et al. (2005). Cloning and characterization of HIV-1-inducible astrocyte elevated gene-1, AEG-1. Gene. 353(1): 8-15.
- Lee SG, Kim K, Kegelman TP, Dash R, et al. (2011). Oncogene AEG-1 promotes Glioma-induced neurodegeneration by increasing glutamate excitotoxicity. Cancer Res.71 (20): 6514-6523.
- O'Bryan JP, Frye RA, Cogswell PC, Neubauer A, et al. (1991). axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biology. 11 (10): 5016-5031.
- Davidsen KT, Haaland GS, Lie MK, Lorens JB et al. (2017). the Role of Axl Receptor Tyrosine Kinase in Tumor Cell Plasticity and Therapy Resistance. In: Akslen L, Watnick R. (eds). Biomarkers of the Tumor Microenvironment. Springer: Cham. 351-376.
- Miller MA, Oudin MJ, Sullivan RJ, Wang SJ, et al. (2016). Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase InhiBitor Resistance. Canc Discovery. 6 (4): 382-399.
- Gay CM, Balaji K and Byers LA. (2017). Giving AXL the axe: targeting AXL in human malignancy. Brit J Cancer. 116 (4): 415-423.
- Hutterer M, Knyazev P, Abate A, Reschke M, et al. (2008). Axl and growth arrest specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin Cancer Res. 14(1): 130-138.
- Vajkoczy P, Knyazev P, Kunkel A, Capelle HH, et al. (2006). Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc Natl Acad Sci USA. 103(15): 5799-57804.
- Keating AK, Kim GK, Jones AE, Donson AM, et al. (2010). Inhibition of Mer and Axl Receptor Tyrosine Kinases in Astrocytoma Cells Leads to Increased Apoptosis and Improved Chemosensitivity. Mol Cancer Ther. 9(5): 1298- 1307.
- Bogler O, Huang HJ, Kleihues P and Cavenee WK. (1995). The p53 gene and its role in human brain tumors. Glia 15(3): 308-327.
- Hainaut P, Butcher S and Milner J. (1995). Temperature sensitivity for conformation is an intrinsic property of wild-type p53. Br J Cancer 71(2): 227-231.
- Ohgaki H and Kleihues P. (2005). Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 64(6): 479-489.
- Ohgaki H, Dessen P, Jourde B, Horstmann S, et al. (2004). Genetic pathways to glioblastoma: a population-based study. Cancer Res. 64(19): 6892-6899.
- Verhaak RG, Hoadley KA, Purdom E, Wang V, et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 17(1): 98-110.
- Bello MJ, Alonso ME, Aminoso C, Anselmo NP, et al. (2004). Hypermethylation of the DNA repair gene MGMT: association with TP53 G: C to A: T transitions in a series of 469 nervous system tumors. Mutat Res. 554(1-2): 23-32.
- El-Husseini AE, Fretier P and Vincent SR. (2001). Cloning and characterization of a gene (RNF22) encoding a novel brain expressed ring finger protein (BERP) that maps to human chromosome 11p15.5. Genomics. 71(13): 363- 367.
- Hatakeyama S. (2011). TRIM proteins and cancer. Nature reviews. Cancer.11 (11): 792-804.
- Chen G, Kong J, Tucker-Burden C, Anand M, et al. (2014). Human Brat Ortholog TRIM3 Is a Tumor Suppressor That Regulates Asymmetric Cell Division in Glioblastoma. Cancer Res. 74(16): 4536-4548.
- Ichimura K, Schmidt EE, Miyakawa A, Goike HM, et al. (1998). Distinct patterns of deletion on 10p and 10q suggest involvement of multiple tumor suppressor genes in the development of astrocytic gliomas of different malignancy grades. Gen Chromos Cancer. 22(1): 9-15.
- Fults D, Pedone CA, Thompson GE, Uchiyama CM, et al. (1998). Micro- satellite deletion mapping on chromosome 10q and mutation analysis of MMAC1, FAS, and MXI1 in human glioblastoma multiforme. Int J Oncol. 12(4): 905- 910.
- Leenstra S, Oskam NT, Bijleveld EH, Bosch DA, et al. (1998). Genetic sub-types of human malignant astrocytoma correlate with survival. Int J Cancer. 79(2): 159-165.
- Nakamura M, Ishida E, Shimada K, Kishi M, et al. (2005). Frequent LOH on 22q12.3 and TIMP-3 inactivation occur in the progression to secondary glioblastomas. Lab Invest. 85(2): 165-175.
- von Deimling A, Nagel J and Bender B. (1994). Deletion mapping of chromosome 19 in human gliomas. Int J Cancer. 57(5): 676-680
- Smith JS, Alderete B and Minn Y. (1999). Localization of common deletion regions on 1p and 19q in human gliomas and their association with histological subtype. Oncogene. 18(28): 4144-4152.
- Bello MJ, Vaquero J, de Campos JM, Kusak ME, et al. (1994). Molecular analysis of chromosome 1 abnormalities in human gliomas reveals frequent loss of 1p in oligodendroglial tumors. Int J Cancer. 57(2): 172-175.
- Bello MJ, Leone PE, Nebreda P, de Campos JM, et al. (1995). Allelic status of chromosome 1 in neoplasms of the nervous system. Cancer Genet Cytogenet. 83(2): 160- 164.
- Kaghad M, Bonnet H, Yang A, Creancier L, et al. (1997). Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 90(4): 809-819.
- Lees JA, Saito M, Vidal M, Valentine M, et al. (1993). The retinoblastoma protein binds to a family of E2F transcription factors. Mol Cell Biol. 13(12): 7813-7825.
- Kwon BS, Tan KB, Ni J, Oh KO, et al. (1997). A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J Biol Chem. 272(22): 14272- 14276.
- Kemper O, Derre J, Cherif D, Engelmann H, et al. (1991). The gene for the type II (p75) tumor necrosis factor receptor (TNF-RII) is localized on band 1p36.2-36.3. Hum Genet. 87(5): 623-624.
- Bodmer JL, Burns K, Schneider P, Hofmann K, et al. (1997). TRAMP, a novel apoptosis-mediating receptor with sequence homology to tumor necrosis factor receptor 1 and Fas (Apo-1/CD95). Immunity. 6(1): 79-88.
- Wu GS, Burns TF, McDonald ER, Jiang W, et al. (1997). KILLER/DR5 is a DNA damage-inducible p53 regulated death receptor gene. Nat Genet. 17(2): 141-143.
- Rasio D, Murakumo Y, Robbins D, Roth T, et al. (1997). Characterization of the human homologue of RAD54: A gene located on chromosome 1p32 at a region of high loss of heterozygosity in breast tumors. Cancer Res. 57(12): 2378-2383.
- Lapointe J, Lachance Y, Labrie Y and Labrie C. (1996). A p18 mutant defective in CDK6 binding in human breast cancer cells. Cancer Res. 56(20): 4586-4589.
- Kraus JA, Koopmann J, Kaskel P, Maintz D, et al. (1995). Shared allelic losses on chromosomes 1p and 19q suggest a common origin of oligodendroglioma and oligoastroytoma. J Neuropathol Exp Neurol. 54(1): 91-95.
- Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, et al. (1998). Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligo dendrogliomas. J Natl Cancer Inst. 90(19): 1473-1479.
- Ueki K, Ono Y, Henson JW, Efird JT, et al. (1996). CDKN2/ p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res. 56(1): 150- 153.
- Burns KL, Ueki K, Jhung SL, Koh J, et al. (1998). Molecular genetic correlates of p16, cdk4, and pRb immunohistochemistry in glioblastomas. J Neuropathol ExpNeurol. 57(2): 122-130.
- Friend SH, Bernards R, Rogelj S, Weinberg RA, et al. (1986). A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 323(6089): 643-646.
- Collins N, McManus R, Wooster R, Mangion J, et al. (1995). Consistent loss of the wild type allele in breast cancers from a family linked to the BRCA2 gene on chromosome 3q12-13. Oncogen. 10: 1673-1675.
- Gudmundsson J, Johannesdottir G, Bergthorsson JT, Arason A, et al. (1995). Different tumor types from BRCA2 carriers show wild-type chromosome deletions on 13q12- q13. Cancer Res. 55(21): 4830-4832.
- Tsuzuki T, Tsunoda S, Sakaki T, Konishi N, et al. (1996). Alterations of retinoblastoma, p53, p16 (CDKN2), and p15 genes in human astrocytomas. Cancer. 78(2): 287-293.