Information Links
Related Conferences
Previous Issues Volume 8, Issue 1 - 2023
Liver Cirrhosis & Hepatocellular Carcinoma in Chronic Hepatitis B: Role of Hepatic Vena Cava Syndrome in the Pathogenesis
Santosh Man Shrestha*
Liver Foundation Nepal, Kathmandu, Nepal
*Corresponding author: Dr. Santosh Man Shrestha, MBBS, FRSTM &H, FRCPE, Liver Foundation Nepal, Sitapaela Height, Nagarjun 4, PO Box # 13273-Kathmandu, Nepal. Tel: 977-9841237627; E-mail: [email protected].
Received Date: March 15, 2023
Published Date: April 18, 2023
Citation: Shrestha SM. (2023). Liver Cirrhosis & Hepatocellular Carcinoma in Chronic Hepatitis B: Role of Hepatic Vena Cava Syndrome in the Pathogenesis. Mathews J Gastroenterol Hepatol. 8(1):19.
Copyrights: Shrestha SM. © (2023).
ABSTRACT
Background: Chronic hepatitis B (CHB) is highly prevalent in some Afro-Asian countries and is associated with high incidence of liver cirrhosis (LC) and hepatocellular carcinoma (HCC), whereas in the West with low prevalence it has a benign course. Pathogenesis of LC in CHB is not well-defined. Materials & Method: Hepatic vena cava syndrome (HVCS), a disease of hepatic venous outflow obstruction is a comorbid condition of CHB patients in Nepal. Presence of HVCS is ascertained by ultrasonography and color Doppler (US/CD) examination of inferior vena cava and liver. This is a retrospective study of a 1542 CHB patients followed for a long period to assess its clinical course. Of these, 988 patients were categorized into two groups based on assay of HBeAg: HBeAg-positive 19% into replicative phase, HBeAg-negative 81% in non-replicative phase. Results: Eighty per cent of the patients were asymptomatic at the time of diagnosis. Acute exacerbations (AE) developed precipitated by bacterial infection with elevation of serum aminotransferases in 80%, and in 11.6 % it was followed by ascites with US/CD evidence of HVOO. About 14% developed mild splenomegaly with hematological features of hypersplenism. These features were consistent with natural history of HVCS. LC developed in 21.3% and HCC in 4.7 % of patient. The incidences of LC and HCC in replicative and non-replicative phases of CHB were similar. But patients with LC and HCC had high incidence of AEs, ascites and hypersplenism. Conclusion: HVCS contributed to the symptomatic clinical course and development of LC and HCC in CHB.
Keywords: Chronic Hepatitis B, Hepatic Vena Cava Syndrome, Hepatic Venous Outflow Obstruction, Hypersplenism, Cirrhosis, Hepatocellular Carcinoma.
ABBREVIATIONS
AE: acute exacerbation; Anti-HBc: Antibody to Hepatitis B Core Antigen; CHB: Chronic Hepatitis B; CAH: Chronic Active Hepatitis; CPH: Chronic Persistent Hepatitis; HV: Hepatic Vein: HBsAg: Hepatitis B Surface Antigen; HBeAg: Hepatitis B e Antigen; HBcAg: Hepatitis B Core Antigen; HCC: Hepatocellular Carcinoma; HVOO: Hepatic Venous Outflow Obstruction; IVC: Inferior Vena Cava; LC: Liver Cirrhosis; US/CD: Ultrasonography and Color Doppler.
INTRODUCTION
Hepatitis B virus (HBV) infects about 2 billion people worldwide, most of who recover spontaneously without any sequel. Chronic hepatitis B (CHB) develops in about 5% of the infected persons. There are about 400 million persons with CHB in the world 80% of who are in Asia and Africa [1]. Based on epidemiologic and clinical studies in these countries HBV is considered a common cause of liver cirrhosis (LC) and hepatocellular carcinoma (HCC) in man [2,3].
Chronic hepatitis B develops in persons in whom the virus succeeds in evading the host immune system. The incidence of CHB is inversely related to the age when the infection is acquired, being highest when the infection is acquired in infancy by vertical transmission. Mechanisms evolved by HBV to overcome the host immune system included production of large amounts of antigens HBsAg and HBeAg [4]. HBeAg is produced by the core gene that also produces HBcAg. HBcAg is the viral antigen targeted by host’s cytotoxic T lymphocyte the immunocytes responsible for clearance of the HBV infected hepatocytes. Antibody against HBcAg does not cross-react with HBeAg, but HBeAg reacts with HBcAg specific cytotoxic T cell receptor. Over production of HBeAg is thus an important strategy of the virus to block the host immune response [5]. Large amount of HBsAg produced by the virus swamps the protective antibody produced by the host preventing its elimination and allowing it to circulate freely in the body [4].
HBV has a DNA genome but it replicates in the liver by transcribing RNA copies continuously in the infected cells [6]. RNA genomes are used as templates to produce DNA genomes by reverse transcription. The process of viral replication in the hepatocyte to viral secretion is regulated in such a manner that it does not escalate to a level harmful to the cell. A very high level of HBV DNA is compatible with normally functioning hepatocytes. HBV is not a cytopathic virus [7,8].
Chronically infected persons often remain asymptomatic throughout life. Natural history of CHB is divided into three viral phases. The early replicative phase is characterized by presence of HBeAg, rapid multiplication of the virus in the hepatocytes and a very high HBV DNA level in the blood. It is followed by non-replicative phase where HBeAg become negative with decline in viral replication. When anti-HBe becomes positive HBV DNA level becomes very low. Both these phases last for several decades. Serum and liver HBsAg bear an inverse relation to each other during the evolution of chronic hepatitis B infection and the quantity of HBsAg in serum declines but in tissue rises gradually with time [9]. Large numbers of HBsAg-positive hepatocytes are observed in long standing healthy carriers and patients with cirrhosis. The pattern and localization of HBV antigens in liver shows no correlation to cell necrosis or damage [7]. Control of the infection with sero-clearance of HBsAg is followed by development of anti-HBs. HBV DNA may remain positive even after clearance of HBsAg. CHB thus often is a life-long infection in some.
There is a geographic variation in the clinical course and outcome of CHB. In Europe and North America patients remain asymptomatic with normal transaminases level with little or no occurrence of LC and HCC [10,11] and the condition is labeled “healthy carrier”. The course of HBV carriers in Japan is also uneventful throughout, where 75% achieved seroconversion to anti-HBe before adulthood [12]. Some HBeAg positive carriers develop small bouts of mild ALT elevations that last for a few months and a subpopulation of CHB especially those who remain HBeAg positive after 30 years have risk of developing LC and HCC at around 50 years age. In India CHB patients exhibited mild transaminases elevation and abnormal liver biopsy findings in high proportion both in the replicative and non-replicative phases [13] and low incidence of HCC [14]. CHB in Taiwan and China begins asymptomatically and later develops bouts of acute exacerbations with high incidence of both LC and HCC [15,16]. Factors determining the clinical course and outcome in CHB are poorly understood. Elevated transaminases levels during HBeAg seroconversion when immune-clearance of infected hepatocytes occur is related to CHB, but its occurrence during replicative, non-replicative phases and after control of infection is not well understood. A prospective study in Taiwan showed that person with HBeAg seropositivity with repeated episodes of AEs and hepatic decompensation had high incidence of LC [17]. In contrast in Italy CHB patient positive for anti-HBe with chronic active hepatitis (CAH) are known to develop LC [18]. LC in CHB is considered to develop through chronic hepatitis. Histological changes observed in liver biopsies in CHB however were frequently non-specific or chronic persistent hepatitis (CPH) which rarely progressed to more serious liver disease at follow up [19]. A prospective study of 292 children with CHB where liver biopsies at presentation showed moderate CAH in 127, severe CAH in 39, CPH in 102 and 14 lobular hepatitis none progressed to cirrhosis during 10 year follow-up [20]. Thus outcome based on chronic hepatitis is unpredictable and pathogenesis of LC in CHB remains poorly understood.
Hepatitis in intravenous drug abusers was associated with persistence of inflammatory changes in the liver even after HBsAg seroconversion and appearance of anti-HBs, which led authors to conclude that these changes were not related to HBV but perhaps to some comorbid condition/s [21]. Drug abusers are prone to develop HCV and bacterial infections from dirty syringes. In the West alcoholic patients have higher exposure rate to HBV than general population [22], but presence of HBV antibodies had no effect in the outcome of alcoholic liver disease [23].
Nepal has low prevalence of CHB with HBsAg rate of 0.9% [24]. The infection is acquired predominantly during adolescent age group by horizontal transmission [25]. One small ethnic group with high incidence of the infection and spread by vertical transmission was identified [26]. LC and HCC were common [27-29] and 56% of these were positive for HBV DNA and 12% for HCV RNA [29]. Hepatic vena cava syndrome (HVCS) a disease of inferior vena cava (IVC) that causes recurrent hepatic venous outflow obstruction (HVOO) is endemic in Nepal [30] and is associated with high incidence of LC and moderate incidence of HCC [31]. HVCS was a common comorbid condition of CHB patients with LC and HCC [29].
Hepatic vena cava syndrome previously labeled ‘membranous obstruction of inferior vena cava (MOVC)’ and considered a congenital vascular anomaly was then managed by surgery or endovascular procedures [32]. The disease was lumped together under Budd-Chiari syndrome with hepatic vein thrombosis related to hypercoagulable conditions [33]. HVCS is now recognized as a separate clinical entity-a bacterial infection induced chronic obliterative disease of inferior vena cava at the site of hepatic vein opening [34-36]. The initial lesion in the disease is a localized thrombophlebitis at the site of hepatic vein opening [37], which on resolution converts to a localized thickened posterior wall or mild stenosis followed by dilatation of the distal segment and development of cava-caval collaterals. Recurrence of bacterial infections resulted in acute exacerbations (AE) with depositions of fresh thrombophlebitis at the site that extended downward along posterior wall of the IVC and into hepatic veins causing HVOO. Large thrombophlebitis formed at the site of hepatic vein opening during acute stage or AE resulted in ascites occasionally associated with pleural effusion from severe HVOO. Ascites in HVCS is characterized by simultaneous development of bacterial peritonitis [38].
Organization and fibrosis of the thrombophlebitis results in intimal thickening increase in stenosis or development of a membrane in IVC and hepatic veins. Development of true membrane in IVC was observed in about 1% of the patients. The deep and superficial collaterals that developed in the disease were described by Pleasants in 1911 [39]. Superficial collaterals are seen as dilated veins over abdomen and back. Because of the extensive network of collaterals patients with HVCS often remains asymptomatic. Patients with recurrent AEs develop splenomegaly with hematological features of hypersplenism, commonly thrombocytopenia and neutropenia [40]. Patients with severe and recurrent AEs develop LC and HCC [31].
HVCS affects people of all age groups from infancy to old age. Its prevalence in a community is inversely related to the standard of hygienic and nutrition [30,41]. It is diagnosed by ultrasonography and color Doppler (US/CD) examination of IVC and the liver [42]. Common findings are localized stenosis of the IVC with thickened posterior wall at the site of hepatic vein opening, presence of old organized thrombophlebitis of different ages along the posterior wall of distal dilated segment often with evidence of hepatic venous outflow obstruction (Fig 1a & 1b) and abnormal blood flow pattern, biphasic or continuous instead of triphasic in IVC. HVCS has a long asymptomatic course with occasional acute exacerbation precipitated by bacterial infection marked by mild elevations of aminotransferases and/or bilirubin or ascites associated with neutrophil leukocytosis and elevation of levels of C-reactive protein and erythrocyte sedimentation rate. The disease is now managed by medical treatment [43].
In Nepal all CHB patients had HVCS as a comorbid condition. This is a retrospective study of 1542 patients of CHB seen over 22 year period from 1998 to 2020 many of whom had been followed for long period to assess its clinical course.
MATERIALS AND METHODS
HBsAg was detected during check-up for employment, blood donation or family-check. At the time of diagnosis 80% of CHB patients were asymptomatic. Some presented with mild jaundice and/or elevation of transaminases. Blood test done in HBsAg positive include anti-HBc, HBeAg, anti-HBe. Patients were categorized based on HBeAg assay in 969; into HBeAg positive 184 (19%) in replicative phase, 785 (81%) HBeAg negative in non-replicative phase, HBeAg was not assayed in 573 patients and it formed the 3d group. HBV viral load was assayed 484 times in a total of 305 patients that included 70 in replicative phase, 235 in non-replicative phase. Viral load > 20,000 IU/ml was considered high, 2000 to 20,000 IU/ml as medium and < 2000 IU/ml as low.
As hepatic vena cava syndrome is endemic in the country US/CD examination of inferior vena cava and liver was done routinely and all showed presence of HVCS. Diagnosis of HVCS was confirmed by inferior vena cavogram and/or liver biopsy in patients seen before 2000. Presence of risk factors of other liver diseases-alcohol was assayed by history and hepatitis C by blood test for anti-HCV and HCV RNA. HCV RNA was positive in 9 out of 717 tested and history of significant alcohol intake defined as > 40 g daily for at least 10 years was obtained in 99 (6.4%) patients.
Routine blood tests at presentation and during follow-up included-hematology (total and differential WBC count, platelet count, hemoglobin and ESR), liver (ALT, AST, alkaline phosphates, protein and albumin) and renal (urea, creatinine and electrolyte) tests and assay of C-reactive protein (CRP). Blood culture for aerobic organisms was done once in 206 patients that presented with fever. Presence of ascites was ascertained by ultrasonography. Assay of ascitic fluid and its inoculation for aerobic culture were done in 80 patients as described earlier [38]. Hypersplenism was diagnosed in patient with platelets count < 150,000/µ L, and/or WBC count < 4000/µ L with/without anemia (hemoglobin < 10g/dL). Diagnosis of cirrhosis was also based on US/CD examination that showed coarse echotexture of liver parenchyma with irregular surface and rounded edge. It was confirmed by biopsy in 20 and by direct inspection and biopsy in four, 2 during splenectomy for hypersplenism and 2 during cholecystectomy for gall stone. HCC was diagnosed on detection of space-occupying lesion) with high alpha fetoprotein level. It was confirmed by biopsy in four and in others by CT scan and FNAC. In two patients who presented to a surgeon with upper abdomen pain and swelling, the tumor was detected at surgery and diagnosis of HCC was confirmed by wedge biopsy.
After 2005 patients were advised to have blood tests for total and differential WBC count, ESR, CRP, bilirubin, ALT and AST any time they developed fever or suspected bacterial infection or after any invasive procedure and to take oral antibiotic if CRP was elevated. Acute exacerbation was treated with oral antibiotic till CRP returned to normal. Patients with ascites also had salt restricted diet and diuretic/s. One hundred twenty patients received antiviral treatment mainly lamivudine or entecavir for long period. Ten patients seen at early period received interferon or pegylated interferon. Patients were seen with AEs and at 6 month to 1 yearly interval. Follow up period were 6 months-1 year in 307, 1 to 5 years in 329, 6 to10 years in 145, and 11 to 30 years in 96.
RESULTS
CHB patients were predominantly (76%) male and young (20-40 years age) and 81% were in non-replicative phase at the time of diagnosis. Follow up showed that the replicative phase usually lasted for 1-2 decades and non-replicative phase for about 2 ½ decades. In a very few who acquired the infection by vertical transmission replicative phase with very high HBV DNA continued to/beyond 5th decade. A total of 47 patients (3%) achieved control of the infection around 5th decade with loss of HBsAg but were still positive for HBV DNA. The HBV viral load was high in patients in replicative phase and low in 69 % of patients in non-replicative phase.
Patients had long asymptomatic course. One or more episodes of mild AEs with jaundice and/or elevation of transaminases developed in 80% of the patients. Severe AE with ascites developed in 179 (11.6 %). Color Doppler evidence of HVOO was observed in all patients with ascites and 70% of patients with mild AEs. AE developed in some at early replicative phase, and more frequently at the end of non-replicative phase or after control of infection.
Patients with ascites had a large thrombophlebitis in IVC at the site of hepatic vein ostia (Figure 1c). Ascites was moderate to severe but was minimal and detected at ultrasonography in 19. Ascites was associated pedal edema and dilated superficial veins over abdomen and back in 60 patients, and pleural effusion in 11. Pleural effusion was right sided in 8 and bilateral in 3. One had massive right sided pleural effusion with minimal ascites. Ascites followed fever and jaundice in 58% and diarrhea in 18%, multiple boils in 3 and major surgery in five patients. Ascites was recurrent in 6. Incidence of ascites was higher in patients with history of alcohol abuse. The findings of ascitic fluid assay were as follows: specific gravity 1018 (range 1016-1027), protein 3.9gm (range 3.2-6.5g), albumin 1.2g (range 1.0-1.9), and serum ascites albumin gradient was 1.4 (range 1.3-1.8), WBC count 12,000 (range 320-25,000), neutrophil count 80% (range 59-85%), absolute neutrophil count 1360 (range 96-17,908) and 62% had presence of RBC in the fluid. Culture grew microorganism in 36. Organisms isolated were Escherichia coli in 16, Klebsiella species in 10 and Staphylococcus aureus in 10. Presence of bacterial peritonitis was also ascertained by ultrasonography on detection of free floating particles in ascitic fluid, or adhesions and/or thick layer of perihepatitis. Hypoalbuminemia was common among patients with ascites. Ten patients with ascites seen at early period had diagnostic peritoneoscopy where liver was found congested with dilated lymphatics on the surface from which lymph were seen oozing. Liver biopsy done in 4 such patients showed presence of massive ischemic liver damage. A few patients with ascites developed variceal bleeding or bleeding from other sites as nose, rectum, urinary tract and ecchymosis.
Blood culture for aerobic organisms done once in 206 patients with AE with fever including 46 with ascites and grew organisms in 99. Organisms isolated included E. coli in 55, Klebsiella species in 31 and Staphylococcus aureus in 8 and others in reminder 5. Presence of mild splenomegaly was also ascertained by ultrasonography. Mild splenomegaly was detected in 275 and it was associated hematological feature of hypersplenism commonly thrombocytopenia and neutropenia in 220 (14 %). The incidences of ALT elevation, ascites and hypersplenism were similar in replicative and non-replicative phases (Table 1 & 2).
LC was detected in 329 (21 %), whose median age was 38 years (range 2-79). LC developed in 67% with ascites compared to 17% without ascites. Cirrhosis developed rapidly within a few months after ascites, whereas it developed over several years in patients with recurrent mild AE (Figure 2a & 2b). Patients with cirrhosis had 5.7 X and 7 X higher incidences of ascites and hypersplenism respectively compared to those without cirrhosis (Table 3). Cirrhosis was of micronodular type when seen soon after ascites. Biopsy done 6 months after ascites in a patient who continued to have recurrent AE had mixed nodular cirrhosis. LC was of macronodular type in a young boy with HVCS with recurrent AEs for many years [44]. Recurrence of jaundice and/or transaminases elevation or ascites in patient with established cirrhosis was also due to bacterial infection induced HVOO and not due to hepatic decompensation (Figure 2c).
HCC developed in 77 (5%) patients, in 56 with LC and 21 without. The median age of HCC patients with and without LC were 52 and 56 years (range 27-84 years) respectively and male to female sex ratio 4.4:1. HCC developed in 18.9 % patients with ascites compared to 2.9 % patients without ascites. HCC was frequently associated with US/CD evidence of HVOO (Figure 3a, Figure 3b).The incidences of LC and HCC in the replicative and non-replicative phases were similar (table 2). Patients with HCC had 10 X higher incidences of both ascites and hypersplenism compared to patients without HCC (Table 3).
Antiviral treatment reduced HBV DNA level significantly in all patients except in one child in replicative phase [45]. It however did not prevent recurrences of ALT elevations or development of ascites and LC. In patients with replicative phase the DNA level returned to high level after stoppage of the drug. The incidence of HCC declined from 2005 onward. In first 6 years from 1998 to 2004 there were 57 HCC, and in next 15 years from 2005 to 2020 number of HCC patients was 20. The decline was probably due to pro-active approach to diagnose and treatment of bacterial infection.
DISCUSSION
CHB patients in Nepal were characterized by long asymptomatic course with occasional episodes of elevation of transaminases and/or bilirubin in 80% cases. AE was precipitated by bacterial infection and was associated with deposition thrombophlebitis in inferior vena cava at the site of the lesion close to hepatic vein ostia frequently resulting in obstruction to blood flow in IVC and at hepatic vein ostia (Figure 1a & 1b). Patients with large thrombophlebitis developed ascites (Figure 1c). Some patients, especially those with history of recurrent acute exacerbations developed mild splenomegaly and hematological features of hypersplenism (Figure 1c). The incidence of ALT elevation, ascites and hypersplenism was higher in patients with controlled infection (Table 1, Table 2). Patients who received anti-viral treatment achieved effective suppression of HBV DNA but it did not affect or prevent elevations of transaminases or ascites, as was observed in Chinese patients treated with lamivudine [46].
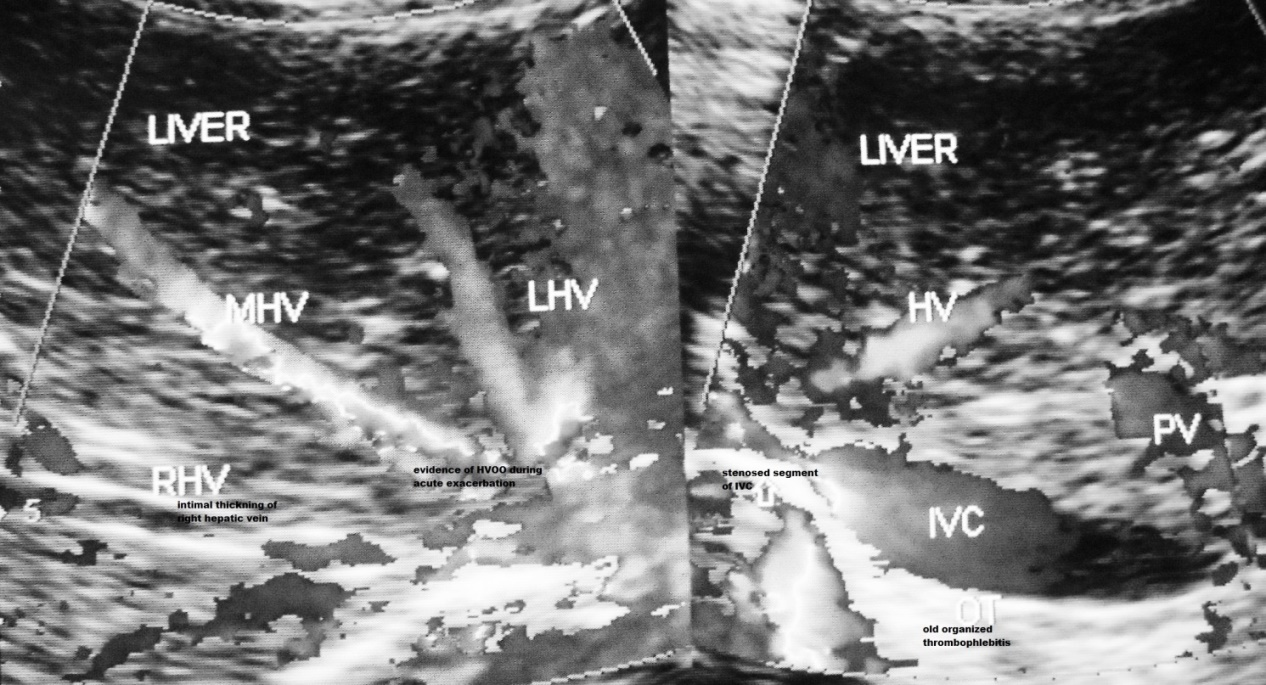
Figure 1a. Hepatic Vena Cava Syndrome is diagnosed by ultrasonography and color Doppler examination of inferior vena cava & liver on detection of localized stenosis at the site of hepatic vein opening. Presence of acute exacerbation is indicated by occurrence hepatic venous outflow obstruction and past AE by presence of old organized thrombophlebitis along posterior wall of distal dilated segment of inferior vena cava.
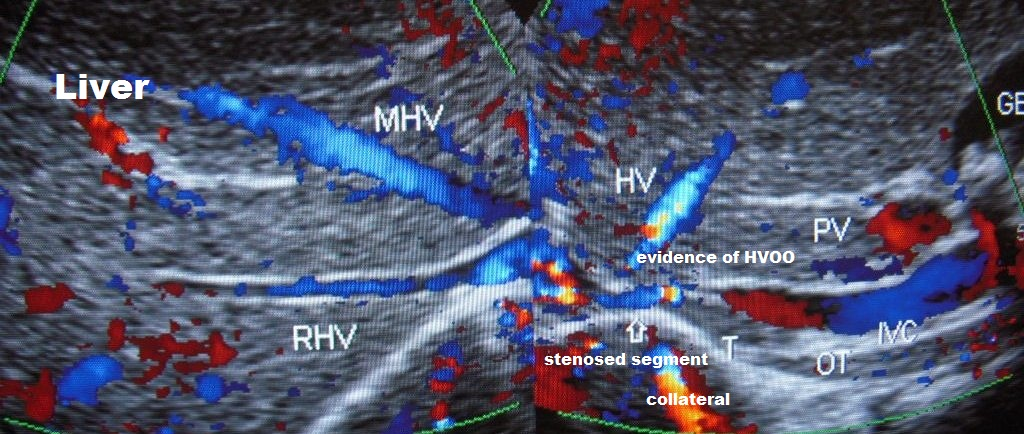
Figure 1b. Ultrasonography and color Doppler evidence of past recurrent episodes of acute exacerbations of hepatic vena cava syndrome: Inferior vena cava shows intimal thickening and stenosis at the site of hepatic vein opening with organized thrombophlebitis of different ages along the posterior wall of the vein. There is evidence of hepatic venous outflow obstruction at the ostia of all major hepatic veins and obstruction to blood flow in IVC at the stenosed segment. Wall of right hepatic vein shows intimal thickening. Liver has uniform increase in echo-texture suggestive of chronic liver disease.
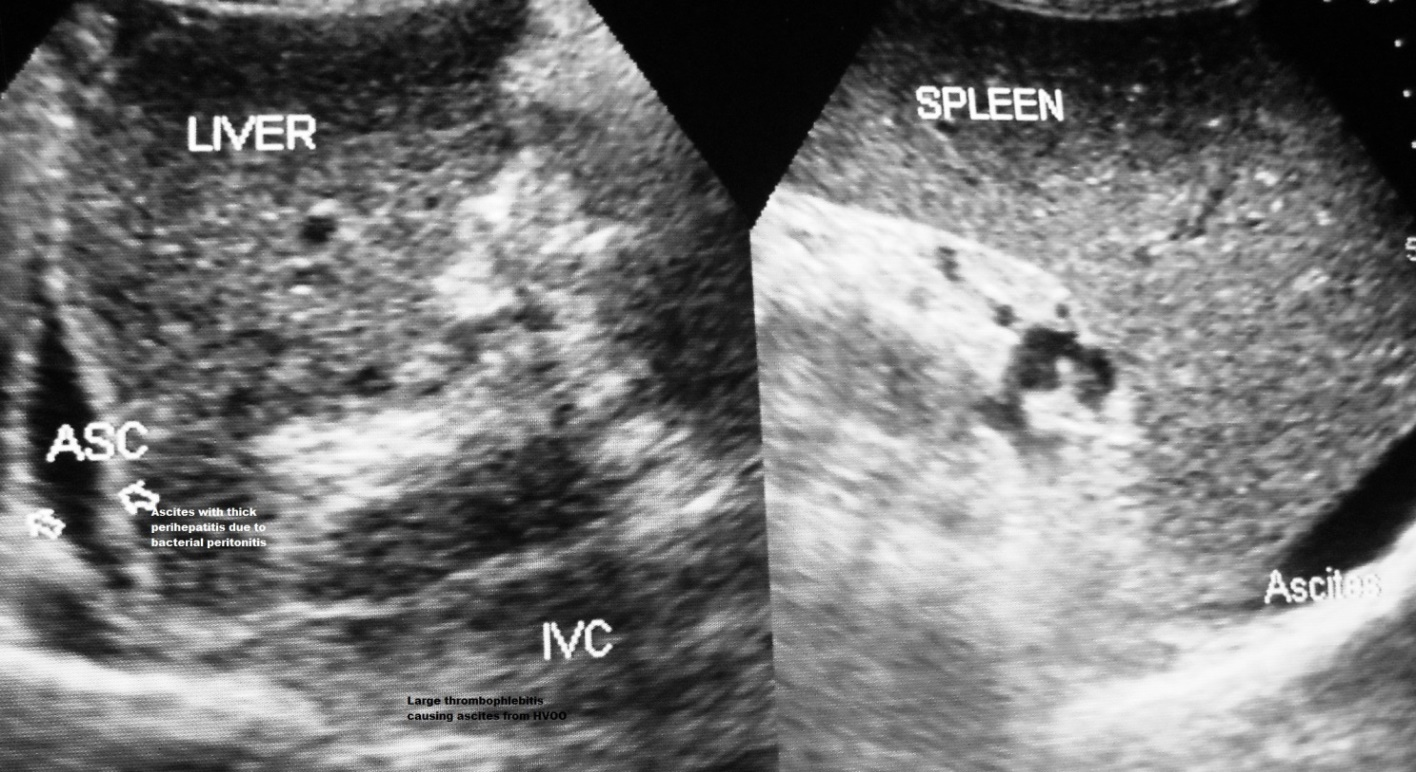
Figure 1c. Development of ascites from severe acute exacerbation: this elderly male patient with CHB in non-replicative phase developed mild jaundice and elevation of transaminases with fever and diarrhea followed two weeks later by ascites. The hepatic portion of inferior vena cava showed intimal thickening, stenosis at the site of hepatic vein opening and is filled with organized thrombophlebitis. Ascites is associated with thick layer of peritoneum indicating presence of bacterial peritonitis. He also has mild splenomegaly from past recurrent acute exacerbations.
Table 1. Incidences of acute exacerbation with elevation of ALT in different phases of Chronic Hepatitis B.
|
Replicative Phase |
Non-replicative |
CHB (HBeAg undetermined) |
Controlled |
|
|
Number of patients |
184 |
785 |
547 |
19 |
|
ALT assay |
||||
|
Normal |
39 (21.4%) |
220 (28.9%) |
33 (6.6%) |
2 (16.5%) |
|
Minimal |
95 (52.1%) |
333 (43.8%) |
233 (46.6%) |
9 (47.3%) |
|
Moderate |
39 (21.4%) |
148 (19.4%) |
152 (30.4%) |
5 (26.3%) |
|
High |
5 (2.7%) |
34 (4.4%) |
47 (9.4%) |
1 (5.2%) |
|
Very High |
4 (2.1%) |
24 (3.1%) |
35 (7.0%) |
2 (10.5%) |
Table 2. Incidence of Ascites, Hypersplenism, Liver Cirrhosis & Hepatocellular Carcinoma in Different Phases of Chronic Hepatitis B.
|
Replicative Phase # 184 |
Non-replicative Phase # 785 |
CHB (no HBeAg assay) # 573 |
Controlled (total) # 47 |
|
|
Ascites |
18 (9.7%) |
78 (9.9%) |
77 (13.4%) |
11 (23.4%) |
|
Hypersplenism |
29 (15.7%) |
135 (17.1%) |
48(8.3%) |
14(29.7%) |
|
LC |
49 (26.6%) |
199 (25.3%) |
48 (8.3%) |
20 (42.5%) |
|
HCC with LC |
4 (2.1%) |
22 (2.8%) |
27 (4.7%) |
3 (6.4%) |
|
HCC |
1 (0.5%) |
3 (0.2%) |
14 (2.4%) |
3 (6.4%) |
Ascites in the past was considered to be developing from decompensation of liver disease [17]. Among our patients ascites developed from severe hepatic venous outflow obstruction due to lesion in IVC at the site of hepatic vein opening. It simulated experimental ascites produced by partial constriction of IVC just above hepatic veins ostia in dogs described by many including Starling [47], Bolton & Bernard [48] and Leavy and Waxler [49]. Severe HVOO results in severe sinusoidal hypertension which precipitated baroreceptor mediated renal sodium retention [49]. The combined effect resulted in rapid oozing of protein rich fluid from the highly porous sinusoid into the space of Disse causing dramatic increase in the formation of the hepatic lymph, most of which is drained by the lymphatic vessel leaving the hilum of the liver that drain into thoracic duct via cysterna chili, but a small spill over from the surface of the liver into peritoneal cavity resulted in ascites. Hyatt, Lawrence and Smith [50] who made direct observation of the surface of the liver in dogs with caval obstruction described appearance of innumerable droplets of fluid over the entire hepatic surface that coalesced and trickled into peritoneal cavity like tears drops resulting in ascites with high protein content. Similar observation was made in our patients with ascites examined by peritoneoscopy.
Sinusoidal hypertension results in reflex reduction of hepatic artery and portal vein inflow [51]. The combined effect resulted in ischemic liver damage around the central hepatic veins as was observed in five patients where liver biopsy was done after ascites. Unlike the immune mediated loss of hepatocytes caused by HBV infection the tissues damaged by ischemia fails to restore to normal and is replaced by lesions of parenchymal extinction that consists of remnants of damaged tissue and elements of repair-fibrosis, neovascularization and arterio-venous shunting [52]. Patient with ascites with severe sinusoidal hypertension developed cirrhosis rapidly within a few months (Figure 2a) [31]. In patients with mild recurrent AE the episodes of ischemic liver damage and regenerative activity around portal tract with formation of parenchymal nodules goes on for long period resulting in the evolution of the lesion over several years to cirrhosis (Figure 2b) [52,53].
HVCS induced cirrhosis at early stage is characterized by presence of a few distinctive US/CD features as dilated hepatic veins, evidence of HVOO, intimal thickening or development of stenosis or membrane or calcified focus in intra-hepatic veins sequel of past thrombophlebitis, edematous or thick wall of gallbladder or a thick layer of perihepatitis, sequel of past ascites with bacterial peritonitis (Figure 2a-2c) [54]. LC with these features that developed with equal frequencies both in replicative and non-replicative phases suggested that LC in CHB was due to HVCS. Further, study of viral replication as monitored by HBeAg and HBV DNA showed that the levels of these had no correlation with the development of chronic liver disease [55]. And a 9 year follow-up study of interferon treatment of Chinese patients showed higher and earlier rates of clearance of HBeAg and HBV DNA but there was no difference in the incidence of hepatic complications between patients and controls and between those who did and did not clear HBeAg (56).
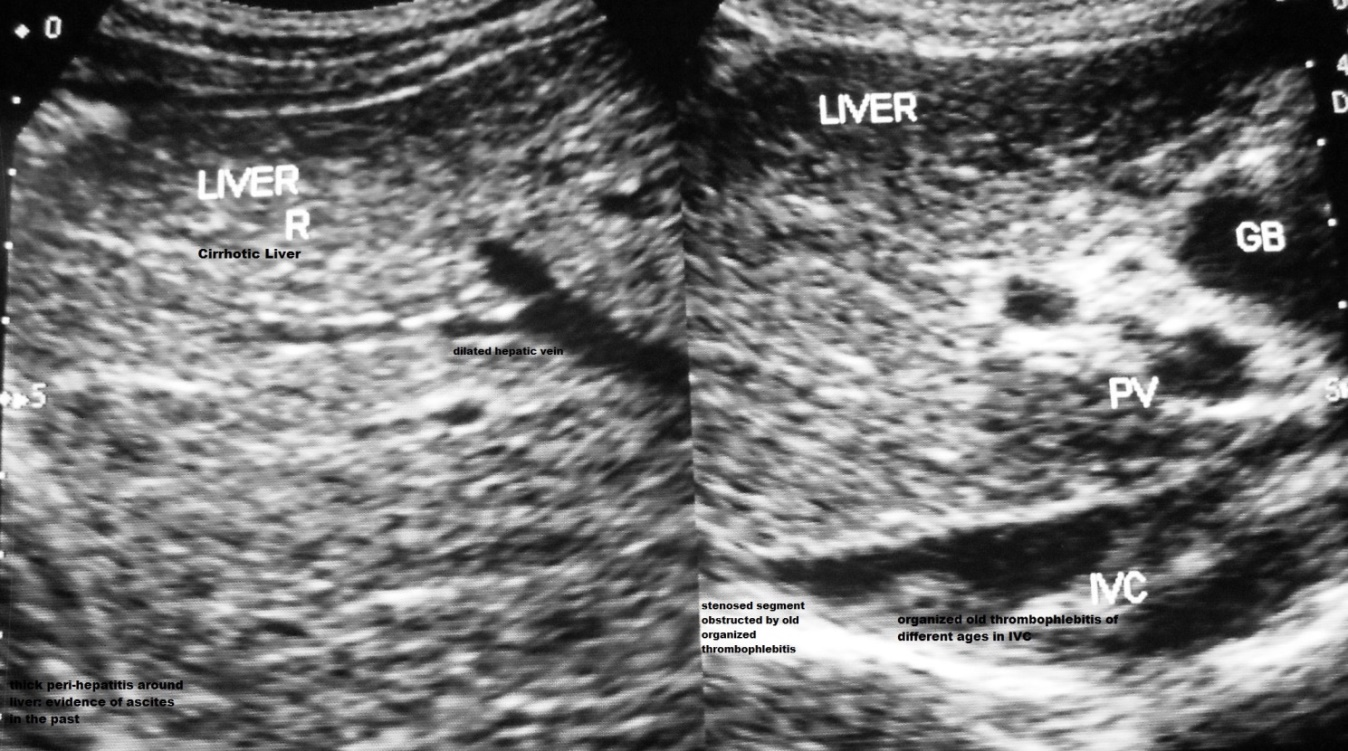
Figure 2a. A 12 year old boy with CHB in replicative phase had recurrent episodes of bacterial infection induced mild acute exacerbations with mild elevations of transaminases. A severe episode of AE with ascites six months back was followed by development of cirrhosis. IVC shows stenosis at the site of hepatic vein opening, old organized thrombophlebitis of different ages along the posterior wall of distal dilated segment. There is a thick layer of perihepatitis from past ascites with bacterial peritonitis. GB wall is thick.
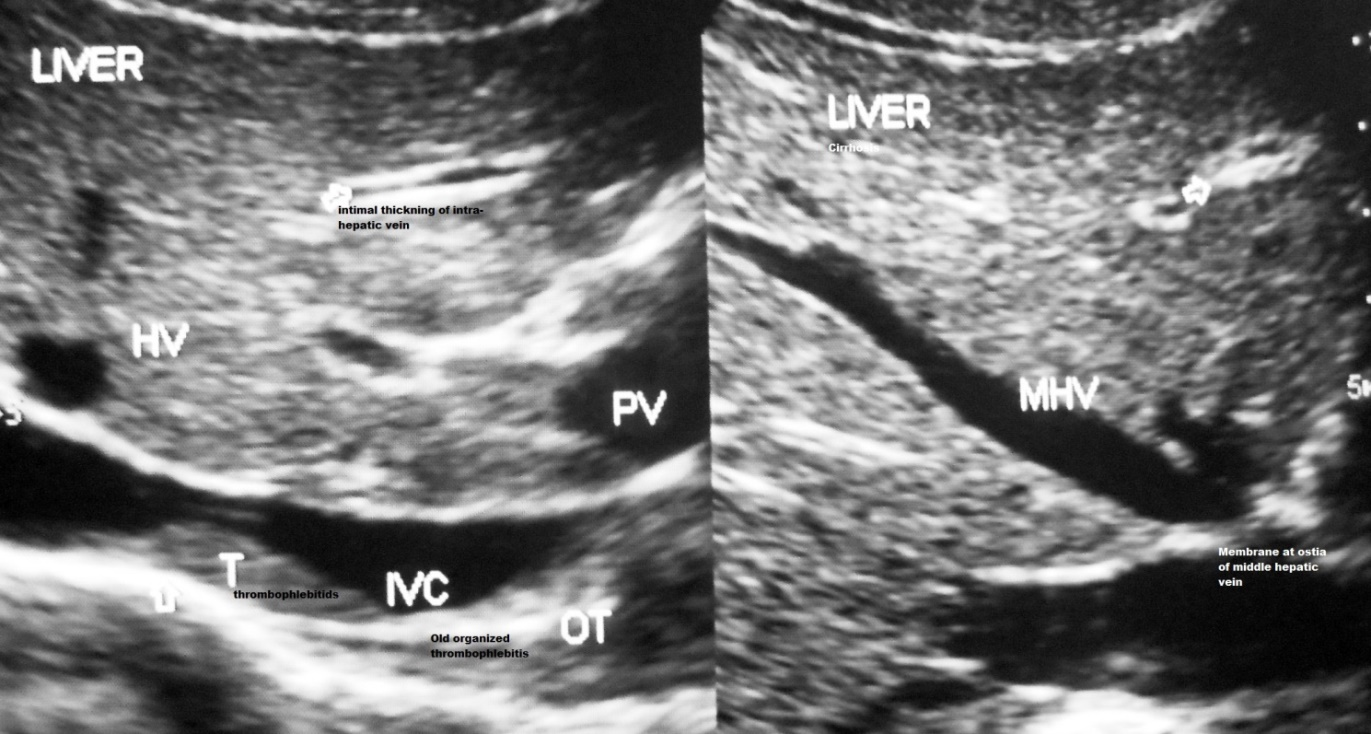
Figure 2b. Diabetic young man with CHB in non-replicative phase developed cirrhosis after recurrent episodes of acute exacerbations over several years period. Inferior vena cava shows intimal thickening, mild stenosis at the site of hepatic vein opening, and multiple old organized thrombophlebitis along the posterior wall. Hepatic veins are dilated; there is a membrane at common ostia of middle and left hepatic veins and intimal thickening of many intra-hepatic veins indicative of past recurrent episodes of AEs.
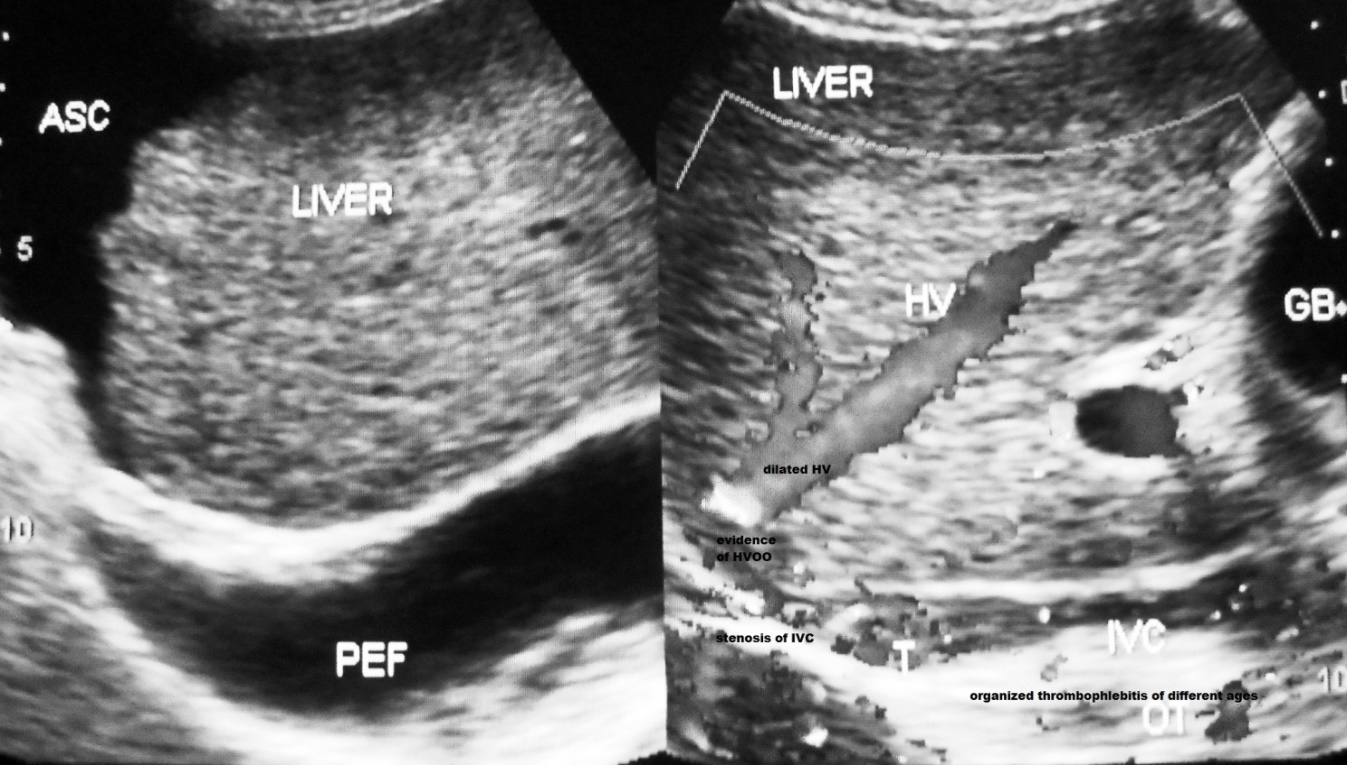
Figure 2c. CHB patients in non-replicative phase with liver cirrhosis developed ascites and pleural effusion from severe hepatic venous outflow obstruction within 10 days of operation for benign enlargement of prostate. US/CD shows dilated hepatic vein with obstruction at its outlet. IVC shows stenosis at the site of hepatic vein opening with obstruction to blood flow by recent thrombophlebitis. The distal dilated segment of the vein shows old organized thrombophlebitis along the posterior wall.
Presence of swollen ground glass hepatocytes demonstrated by orcein stain observed in these patients thus should not be interpreted that the cirrhosis was due to CHB. Orcein positive hepatocyte was due to the presence of hepatitis B surface antigen in the liver cells and it was observed in carriers who had cirrhosis from other causes [57]. Similarly, enlarged portal areas with infiltration of inflammatory cell and pericellular fibrosis occurred in HVCS and its presence should not be interpreted as chronic hepatitis. Histological study of a large cohort of HVCS patients in Nepal showed a variety of changes (Figure 4) that included perivenular and periportal fibrosis that extended into parenchyma in arachnoid fashion in 20, distortion of lobular architecture with widespread fibrosis and occasional regenerative nodule formation in 47, centrilobular congestion and hemorrhage in 18, well developed liver cirrhosis in 12 and presence of hepatocellular carcinoma in the background of dense fibrosis in 7. Liver histology in such patients showed mixed features of cirrhosis with necrosis [58] and should not be interpreted as piecemeal necrosis of chronic hepatitis.
Association between CHB and HCC was considered well established based on epidemiological, clinical [2,3] and molecular studies. Presence of integrated HBV DNA into the host cellular genome was detected in the HBV related HCC [59]. Experimental studies in transgenic mice showed that HBx protein the product of HBV X gene induced HCC in 80% of the animals [60]. Though the HBx protein was expressed soon after birth, HCC developed in the transgenic mice only at 13 months and thereafter. This and observation that in CHB HCC developed in 60-90% in the background of cirrhosis [19] and at around 50 years age suggested that besides viral other factor/s also was necessary for carcinogenesis. In molecular pathogenesis of HCC disorganization and rearrangement of hepatocellular DNA is considered important [61]. HCC develops, in the setting of recurrent liver cell injury that leads to increased liver cell turnover and the host’s regenerative response that leads to mutations in the hepatocyte. This cardinal event occurred in patients with recurrent ischemic liver damage from HVOO. Patients with HCC in this study had recurrent episodes of HVOO (Figure 3a & 3b) as indicated by the highest incidence of ALT elevation, ascites and hypersplenism (Table 3).
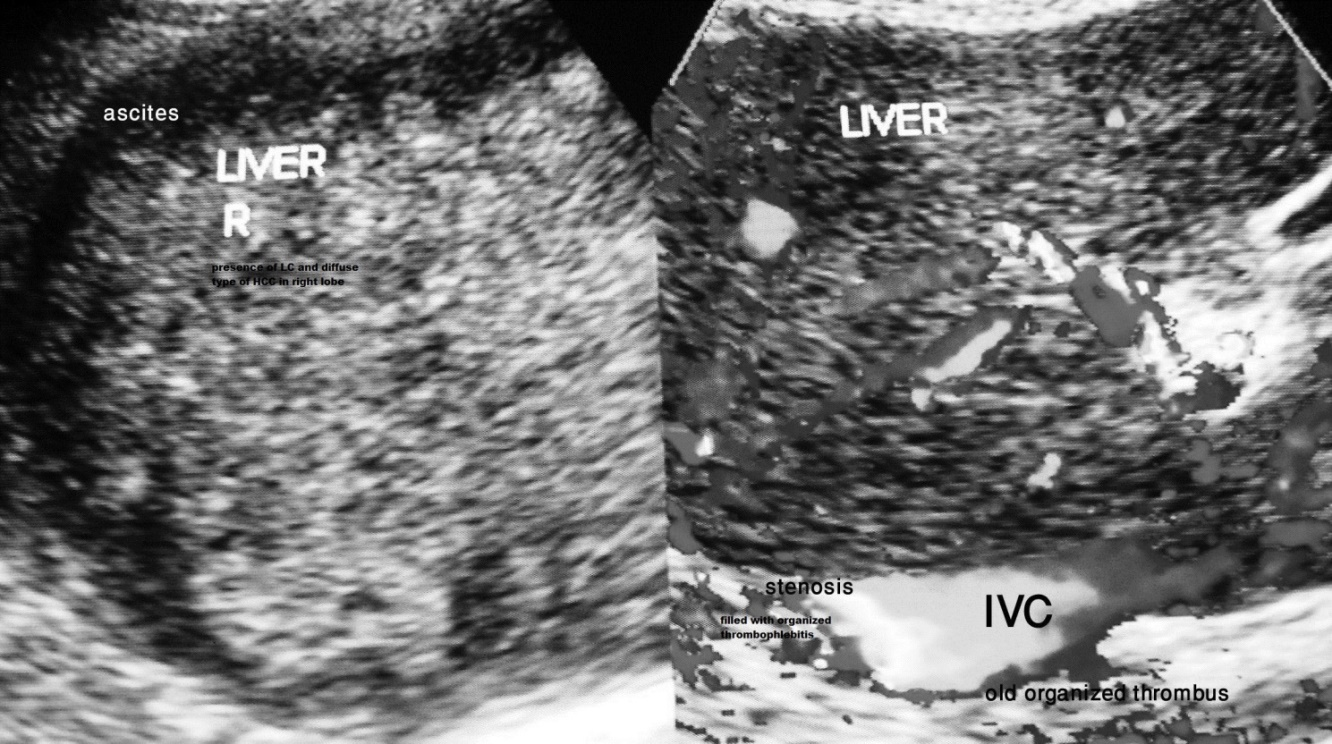
Figure 3a. Ultrasonography and color Doppler of a 57 year old lady that presented with acute upper abdominal pain and vomiting and hemorrhagic ascites. She had dilated superficial veins in right upper abdomen, chest and back in lumbar region. Routine blood test showed HBsAg positive. She was HBeAg negative and anti-HBe positive, HBV DNA level of 2291 IU/ml. Onset of ascites was marked by very high level of C-reactive protein and neutrophil leukocytosis.
US/CD showed marked stenosis of IVC at the site of hepatic vein opening caused by organized thrombophlebitis on its posterior wall resulting in obstruction to blood flow, presence of ascites with evidence of HVOO. Right lobe of liver was replaced by HCC. Its presence was confirmed by liver biopsy and high level of alpha fetoprotein.
She had recurrent pelvic infections in the past. Family check showed that 3 out of her 8 sibs and one son and one elder daughter were HBsAg positive-all in non-replicative phase and asymptomatic. She probably acquired CHB by vertical transmission.
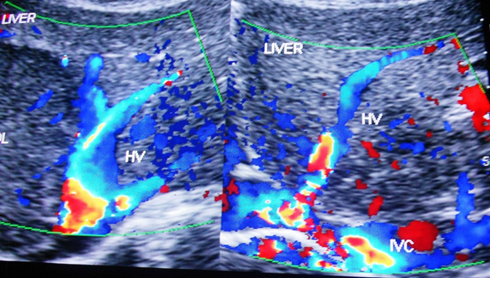
Figure 3b. US/CD of a 50 year old man showing evidence of liver cirrhosis with a large HCC in the right lobe of the liver in a patient with evidence of hepatic vena cava syndrome. IVC shows localized stenosis at the site of hepatic vein opening with evidence of obstruction to blood flow in IVC and hepatic veins.
This patient first presented to a surgeon with upper abdomen pain and swelling. Liver cirrhosis and a large HCC were detected at laparotomy. Diagnosis of HCC was confirmed by liver biopsy and assay for AFP. He was detected HBsAg positive, HBeAg negative and anti-HBe with very low level of HBV DNA.
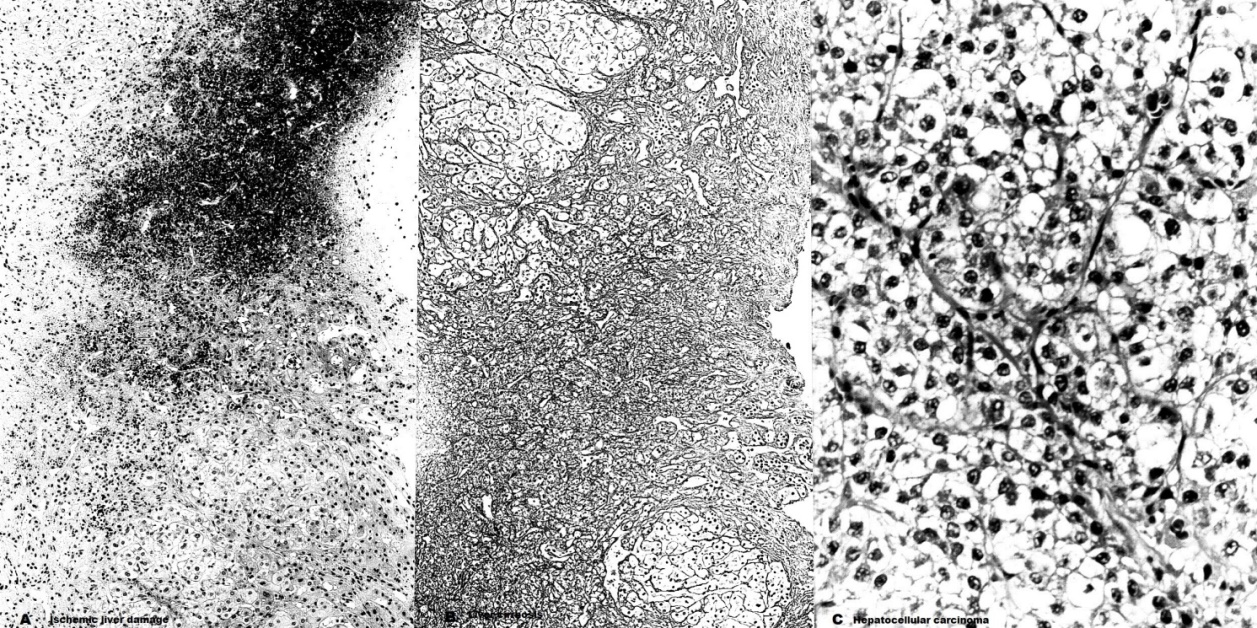
Figure 4. Liver biopsy showing (A) ischemic liver damage from hepatic venous outflow obstruction, HE X 100 (B) liver cirrhosis, silver X 100 & (C) poorly differentiated HCC, X 200.
Table 3. Incidence of Ascites, Hypersplenism, ALT elevation & History of Jaundice in Patients with Chronic Hepatitis B with and without LC & HCC.
|
CHB without LC/HCC |
CHB with LC |
CHB with HCC |
|
|
Number |
1157 |
301 |
77 |
|
Ascites |
53 (4.3%) |
89 (24.9%) |
34 (44.1%) |
|
Hypersplenism |
60 (5.1%) |
124 (41.1%) |
38 (49.3%) |
|
ALT elevation |
272 (23.4%) |
145 (48.1%) |
54 (71.1%) |
|
H/O Jaundice |
215 (18.5%) |
85 (28.2%) |
21 (27.2%) |
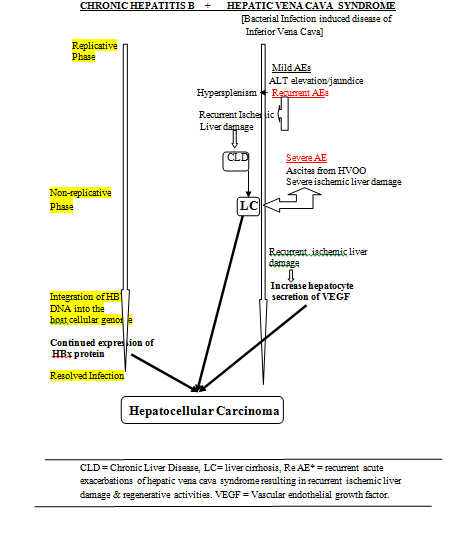
Figure 5. Pathogenesis of LC and HCC in CHB with HVCS as a comorbid condition.
Question arise how frequently HVCS occurred as comorbid condition of CHB in areas with high incidence of HCC. Three-quarters of the patients with CHB in the world are Chinese. Another area of high endemicity is sub-Saharan Africa. These are the very areas from where membranous obstruction of inferior vena cava had been reported [41,64-66]. Wang and his colleagues from China in 2004 reported a large series of 2,564 patients with severe obstruction of IVC managed by surgery or interventional radiological procedures [64], suggesting it as a tip of the iceberg of HVCS in China. Occurrence of HVCS as comorbid condition of CHB among Chinese is further suggested by reports of high incidence of cirrhosis in children with CHB from Taiwan with recurrent mild elevation of transaminases, jaundice and splenomegaly [67]; and of reports of development of cirrhosis in CHB patients from Hong Kong with history of recurrent elevations of ALT, ankle edema, hypoalbuminemia, splenomegaly, and hypersplenism [68]. High incidence of HVCS as a comorbid condition of CHB in South Africa was suggested by high incidence of clinical features as fever (37.5%), jaundice (31.8%), ascites (53.5%) and splenomegaly (39.6%) among 463 patients with HCC of which 56.6% were HBsAg positive reported by Kew [68]. The emphasis in the past on membranous obstruction had resulted in diagnosis of only limited number of cases at late stage frequently after development of HCC [65,66,70,71]. HVCS had occurred in the West in the past [39,72-75], but is now only occasionally reported [75]. The importance of diagnosing HVCS at early stage is illustrated by the case of 62 year old woman with HCC reported by Havlioglu et al [76] from USA that presented with jaundice with elevation of transaminases, hypersplenism with thrombocytopenia, enlarged caudate lobe and dense calcification of the liver capsule. The patient had attended a hospital with liver problem 30 years back where she was treated with a portocaval shunt for esophageal varices. Review of the liver biopsy taken at that time reported as post-necrotic cirrhosis was now found to show changes acute and chronic HVOO consistent with HVCS.
CONCLUSION
CHB patients in Nepal had HVCS as a comorbid condition, both of which had long asymptomatic courses. Occurrence of bacterial infections resulted in acute exacerbations of HVCS marked by elevation of the levels of transaminases followed by ischemic liver damage from HVOO. Recurrent and severe HVOO resulted in development of LC. Long duration of the disease, presence of LC and recurrence of ischemic liver damage from HVOO resulted in development of HCC.
ETHICAL APPROVAL
This retrospective study of patients with chronic hepatitis B was approved by Liver Foundation Research Ethic Committee.
REFERENCES
- WHO. (1996). World Health Report 1996: fighting disease, fostering development. Geneva, Switzerland: World Health organization.
- Beasley RP, Hwang L-Y, Lin C-C, Chen C-S. (1981). Hepatocellular carcinoma and hepatitis B virus: a prospective study of 22707 men in Taiwan. Lancet. 2(8256):1129-1133.
- Okuda K, Beasley RP. (1982). Epidemiology of hepatocellular carcinoma. In: Okuda K, Mackay I, (eds). Hepatocellular carcinoma, Geneva, Switzerland, UICC Report 17. pp. 9-30.
- Blumberg BS. (1994). The hepatitis viruses: accomplishments and problems. In Viral hepatitis and liver disease. In: Nishioka K, Suzuki H, Mishiro S, Oda T, (eds). Springer-Verlag, Tokyo, Japan. p. 7-9.
- Milich DR, McLachlan A, Moriarty A, Thornton GB. (1987). Immune response to hepatitis B core antigen (HBcAg): localization of recognition sites within HBcAg/HBeAg .J Immunol. 139(4):1223-1231.
- Summers J. (1989). The hepadnaviruses: a review of replication and persistence. In: Bianchi L, Gerok W, Maier K P, Deinhardt F, (eds). Infectious Diseases of the Liver. Falk Symposium 54. Chapter 18. Kluwer Academic Publishers, Dordrecht. Netherlands. pp 249-259.
- Peters M, Vierling J, Gershwin ME, Milich D, Chisari FV, Hoofnagle JH. (1991). Immunology and liver. Hepatology. 13(5):977-994.
- Gerber MA. (1987). Immunopathology of chronic hepatitis. In: Farber E, Phillips MJ, Kaufman N, (eds). Pathogenesis of liver diseases. Williams & Wilkins, Baltimore, USA. p. 54-64.
- Lau JYN, Bain VG, Davies SE, Alexander JM, Williams R. (1991). Export of intracellular HBsAg in chronic hepatitis B infection is related to viral replication. Hepatology. 14(3): 416-421.
- Dragosics B, Ferenci P, Hitchman E, Denk H. (1987). Long term follow-up study of asymptomatic HBsAg positive voluntary blood donors in Austria: a clinical and histologic evaluation of 242 cases. Hepatology. 7(2):302-306.
- De Franchis R, Meucci G, Vecchi M, et al. (1993). The natural history of asymptomatic hepatitis B surface antigen carriers. Ann Intern Med. 118(3): 191-194.
- Iino S. (2002). Natural history of hepatitis B & C virus infection. Oncology. 62(suppl 1):18-23.
- Tyagi P, Chauhan P, Kumar R, Sharma BC, Sarin SK. (2004). Normal ALT level do not signify absence of histological injury or significant viremia in incidentally detected asymptomatic HBsAg positive subjects (IDAHS) and its correlation with HBV DNA levels. Ind J Gastroenterol. 23: A56.
- Wang BE, Ma WM, Sulaiman A, Noer S, Sumohajo S, Sumarshidi D, et al. (2002). Demographic, clinical and virological characteristics of hepatocellular carcinoma in Asia: survey of 414 patients from four countries. J Med Virol. 67(3):394-400.
- Chen DS. (1993). Natural history of chronic hepatitis B infection: new light on an old story. J Gastroenterol & Hepatol. 8(5):470-475.
- Lau GKK, Wu PCC, Lai CL. (1997). Natural history of chronic hepatitis B infection. HK Med J. 3(3):283-288.
- Liaw YF, Tai DL, Chu CM, Chen TJ. (1988). The development of cirrhosis in patients with chronic hepatitis B: a prospective study. Hepatology. 8(3):493-496.
- Fattovich G, Brollo L, Alberti A, Pontisso P, Giustina G, Realdi G. (1988). Long-term follow up of anti-HBc positive chronic hepatitis B. Hepatology. 8(6):1651-1654.
- Popper H, Shafritz DA, Hoofnagle JH. (1987). Relation of the hepatitis B virus carrier state to hepatocellular carcinoma. Hepatology. 7(4):764-772.
- Bortolotti F, Calzia R, Cadrobbi P, Giacchini R, Ciraveegna B, Armigliato M, et al. (1986). Liver cirrhosis associated with chronic hepatitis B virus infection in Children. J Pediatr. 108(2):224-227.
- Bianchi L, Zimmerli N, Gudat F. (1979). Viral hepatitis. In: MacSween RNM, Anthony PP, Scheuer PJ, (eds). Pathology of liver. Churchill Livingstone, Edinburgh, Scotland. pp. 164-191.
- Wands JR, Blum HE. (1991). Hepatitis B and C virus and alcohol-induced liver injury. Hepatology. 14(4 Pt 1):730-733.
- Mendenhall CL, Seeff L, Diehl AM, et al. (1991). Antibodies to hepatitis B virus and hepatitis C virus in alcoholic hepatitis and cirrhosis: their prevalence and clinical relevance. Hepatology. 14(4 Pt 1):581-589.
- Shrestha SM. (1990). Seroepidemiology of hepatitis B in Nepal. J Commun Dis. 22(1):27-32.
- Shrestha SM. (2012). Chronic hepatitis B in Nepal: an Asian country with low prevalence of HBV infection. Trop Gastroenterology. 33(2):95-101.
- Shrestha SM, Takeda N, Tsuda F, Okamoto H, Shrestha S, Shrestha VM. (2002). High prevalence of hepatitis B virus infection among Tibetans in Nepal. Trop Gastroenterol. 23(2):63-65.
- Shrestha SM. (1998). Primary liver cancer in Nepal. J Inst Med. 10:177-180.
- Shrestha SM, Tsuda F, Okamoto H, Tokita H, Horikita M, Tanaka T, et al. (1994). Hepatitis B subtypes and hepatitis C virus genotypes in patients with chronic liver disease in Nepal. Hepatology. 19(4):805-809.
- Shrestha SM, Shrestha S, Shrestha A, Tsuda F, Endo K, Takahashi M, et al. (2007). High prevalence of hepatitis B virus infection and inferior vena cava obstruction among patients with liver cirrhosois or hepatocellular carcinoma in Nepal. J Gatroenterol and Hepatol. 22(11):1921-1928.
- Shrestha SM, Okuda K, Uchida T, Maharjan KG, Shrestha S, Joshi BL, et al. (1996). Endemicity and clinical picture of liver disease due to obstruction of the hepatic portion of inferior vena cava in Nepal. J Gastroenterol and Hepatol. 11(2):170-179.
- Shrestha SM. (2009). Liver cirrhosis and hepatocellular carcinoma in hepatic vena cava disease, a liver disease caused by obstruction of inferior vena cava. Hepatol Int. 3(2):392-402.
- Hirooka M, Kimura C. (1970). Membranous obstruction of the hepatic portion of the inferior vena cava. Surgical correction and etiological study. Arch Surgury. 100(6):656-663.
- Janssen HLA, Gaecia-Pagan J-C, Elias E, Mentha G, Hadengue A, Valla D-C. (2003). Budd-Chiari syndrome: a review by an expert panel. J Hepatol. 38(3):364-371.
- Shrestha SM, Kage M, Lee BB. (2017). Hepatic vena cava syndrome: new concept of pathogenesis. Hepatol Res. 47(7):603-615.
- Shrestha SM, Shrestha S. (2007). Hepatic vena cava disease: etiologic relation to bacterial infection. Hepatol Res. 37(3):196-204.
- Shrestha SM. (2017). Hepatic venous outflow obstruction: suggestion of a new classification. J Ren Hepat Disord. 1(2):41-51.
- Shrestha SM, Joshi BL, Shrestha S, Maharjan KG. (2000). Cavographic study of an early stage of obstruction of the hepatic portion of inferior vena cava. J Gastroenterol Hepatol. 15(2):202-210.
- Shrestha SM, Shrestha S. (2002). Bacterial peritonitis in the hepatic vena cava disease: a hypothesis to explain the cause of infection in high protein ascites. Hepatology Res. 24(1):42-49.
- Pleasants JH. (1911). Obstruction of the inferior vena cava with report of eighteen cases. Johns Hopkins Hospital Report. 16:363-548.
- Shrestha SM. (2021). Splenomegaly and hypersplenism in hepatic vena cava syndrome. Hepatology Forum. 2(2):69-75.
- Okuda K. (1982). Memebranous obstruction of the inferior vena cava: Etiology and relation to hepatocellular carcinoma. Gastroenterology. 82(2):376-379.
- Shrestha SM. (2017). Diagnosis of hepatic vena cava syndrome by ultrasonography and color Doppler based on new concept of its pathogenesis. EC Gastroenterology and Digestive System. 2(1):256-270.
- Shrestha SM. (2019). Medical management of hepatic vena cava syndrome (Based on new concept of its pathogenesis). Japanese Journal of Gastroenterology and Hepatology. 1:1-9.
- Shresthha SM, Shrestha S. (2014). Hepatic vena cava syndrome: a common cause of liver cirrhosis in children in Nepal. Trop Gastroenterol. 35(2):85-95.
- Shrestha SM. (2022). Liver Cirrhosis in Chronic Hepatitis B: is it due to Hepatic Vena Cava Syndrome: case reports. J Hum Virol Retrovirol. 9(2):65-73.
- Lai CL, Ching CK, Tung AK, et al. (1997). Lamivudine is effective in suppressing hepatitis b virus DNA in Chinese hepatitis B surface antigen carriers: A placebo-controlled trial. Hepatology. 25(1):241-244.
- Starling EH. (1896). Physiological factors involved in the causation of dropsy. Lancet. 1:1267-1270.
- Bolton C, Barnard WG. (1931). The pathological occurrences in the liver in experimental venous stagnation. J Pathol Bacteriol. 34(6):701-709.
- Leavy M, Waxler MJ. (1987). Sodium retention in dogs with low-grade caval constriction: role of hepatic nerves. Am J Physiol. 253(4 Pt 2):F672-F678.
- Hyatt RE, Lawrence GH, Smith JR. (1955). Observations on the ascites from experimental hepatic congestion. J Lab Clin Med. 45(2):274-280.
- Greenway CV, Stark RD. (1971). Hepatic vascular bed. Phys Rev. 51(1):23-65.
- Wanless IR. (2004). Pathogenesis of cirrhosis. J Gastroenterology and Hepatology. 19(s7): S369-S371.
- Shrestha SM. (2017). Sinusoidal hypertension: a cause of liver cirrhosis in developing countries. Austin J Gastroenterol. 4(3):1-8.
- Shrestha SM. (2015). Liver cirrhosis in hepatic vena cava syndrome (or membranous obstruction of inferior vena cava). World Journal of Hepatology. 7(6):874-884.
- Anderson MG, Murray-Lyon IN. (1985). Natural history of HBsAg carriers. Gut. 26(8): 848-860.
- Lau GKK, Lim WL, Lok ASF. (1997). Nine years follow-up after interferon (IFN) treatment for chronic hepatitis B (CHB) in Chinese patients. Hepatology. 26:259 A.
- Anthony PP, Ishak KG, Nayak NC, Poulsen HE, Scheuer PJ, Sobin LH. (1977). The morphology of cirrhosis: definition, nomenclature, and classification. Bull World Health Organ. 55(4):521-540.
- Singh V, Sinha SK, Naik CK, Bambery P, Kaur U, Verma S, et al. (2000). Budd-Chiari syndrome: Our experience of 71 patients. J Gastroenterol Hepatol. 15(5):550-554.
- Umeda T, Hino O. (2002). Molecular aspects of human hepatocarcinogenesis mediated by inflammation: from hypercarcinogenic state to normo-or hypocarcinogenic state. Oncology. 62(suppl 1):38-40.
- Koike K, Moriya K, Ino S, Yotsuyangi H, Endo Y, Miyamura T, et al. (1994). High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology. 19(4): 810-819.
- Wands JR, Blum HE. (1991). Primary hepatocellular carcinoma. New Eng J Med. 325(10):729-731.
- Ferrara N, Gerber HP, LeCouter J. (2003). The biology of VEGF and its receptors Nature Med. 9(6):669-676.
- Yamaguchi R, Yano H, Iemura A, et al. (1998). Expression of vascular endothelial growth factor in human hepatocellular carcinoma. Hepatology. 28(1):68-77.
- Wang ZG, Zhang FJ, LI XQ, Meng QY. (2004). Management of Budd-Chiari syndrome: what is the best approach? J Gastroenterology and Hepatology. 19(s7): S212-S228.
- Simson IW. (1982). Membranous obstruction of inferior vena cava and hepatocellular carcinoma in South Africa. 82(2):171-178.
- Kew MC, McKnight A, Hodkinson J, Bukofzer S, Esser JD. (1989). The role of membranous obstruction of the inferior vena cava in the etiology of hepatocellular carcinoma in Southern African Blacks. Hepatology. 9(1):121-125.
- Hsu HC, Lin YH, Chang MH, Su IJ, Chen DS. (1988). Pathology of chronic hepatitis B virus infection in children with special reference to intrahepatic expression of hepatitis B virus antigen. Hepatology. 8(2):378-382.
- Lok ASF, Lai CL, Chung HT, Lau JYN, Leung EKY, Wong LSK. (1991). Morbidity and mortality from chronic hepatitis B virus infection in family members of patients with malignant and nonmalignant hepatitis B virus-related chronic liver disease. Hepatology. 13(5):834-837.
- Kew MC. (1989). Hepatocellular Carcinoma with and without Cirrhosis. Gastroenterology. 97(1):136-139.
- Shin SH, Chung YH, Suh DD, Shin JW, Jang MK, Ryu SH, et al. (2004). Characteristic clinical features of hepatocellular carcinoma associated with Budd-Chiari syndrome: evidence of different carcinogenic process from hepatitis B virus associated hepatocellular carcinoma. European J Gastroenterol Hepatol. 16(3):319-324.
- Gown DI, Ko GY, Yoon HK, Sung KB, Kim JH, Lee SS, et al. (2010). Hepatocellular carcinoma associated with membranous obstruction of the inferior vena cava: incidence, characteristics, risk factor and efficacy of TACE. Radiology. 254(2):617-626.
- Osler W. (1879). Obstruction of vena cava inferior and great stenosis of orifices of hepatic veins. J Anat Physiolo. 13(Pt 3):291-304.
- Dixon Man J, Walker Hall I. (1904). Obstruction of the inferior vena cava. Edinb Med J. 16(1):56-62.
- Rigdon RH. (1933). On the relation between thrombophlebitis of inferior vena cava and occlusion of hepatic veins (endophlebitis obliterans). Bull Johns Hopkins Hosp. 53:162-171.
- Rector WCJ, Xu YH, Goldstein I, Peters RL, Reynold TB. (1985). Membranous obstruction of inferior vena cava in the United States. Medicine (Baltimore). 64(2):134-143.
- Havlioglu N, Brunt EM, Bacon BR. (2003). Budd-Chiari syndrome and hepatocellular carcinoma: a case report and review of literature. Am J Gastroenterol. 98(1):201-204.