Previous Issues Volume 3, Issue 1 - 2018
Infantile Hypophosphatasia in Saudi Child. Case Report
Ruba Abo Essa1 , Bothaina Ghazali1 , Bayan Abbag2 , Salman Assiri
1Pediatric department, Abha Maternity and Children's Hospital, Assir, Saudi Arabia.
2Najran University, department of child health, Najran, Saudi Arabia.
Corresponding Author: Ruba Abo Essa, Pediatric department, Abha Maternity and Children's Hospital, Assir, Saudi Arabia. E-Mail: [email protected]
Received Date: 31 Jan 2018 Accepted Date: 26 Feb 2018 Published Date: 02 Feb 2018
Copyright © 2018 Abo Essa R
Citation: Abo Essa R, Ghazali B, Abbag B and Assiri S. (2018). Infantile Hypophosphatasia in Saudi Child. Case Report. Mathews J Pediatr. 3(1): 010.
ABSTRACT
AHypophosphatasia is characterized by low serum alkaline phosphatase (ALP) activity and has a similar radiographic appearance with rickets. There are considerable differences in the severity of the disease. The clinical course of this condition often improves; although early death can occur in the infantile form of the disorder. We experienced one case with severe infantile type of hypophosphatasia.
KEYWORDS
Hypophosphatsia; Hypercalcemia; Calceim; Alkaline Phosphatase; ALP; Rickets.
INTRODUCTION
Hypophosphatasia (HPP) characterized by defective mineralization of bone and dental tissue [1, 2]. The features of this disease include diminished alkaline phosphatase (ALP) activity and increased urinary excretion of Phosphorylethanolamine.
This inborn error of metabolism is a rare inherited form of rickets or osteomalacia due to reduced activity of tissue-nonspecific alkaline phosphatase (TNSALP) caused by loss-of-function mutation within the TNSALP gene marked by low serum ALP [3, 4]. HPP is classified into different clinical types with a remarkable range of severity.
Perinatal HPP is always fatal from profound skeletal hypomineralization [5]. Infantile HPP presents before six months of age with rickets, failure-to-thrive and hypercalcemia occasionally, craniosynostosis or vitamin B6-responsive seizures as well as respiratory failure associated with progressive chest deformity [6, 7]. Childhood HPP features rickets and loss of deciduous teeth, hypoplasia and myopathy while adult HPP causes osteomalacia [7, 8].
Autosomal recessive inheritance accounts for severe HPP while autosomal dominant or recessive transmission underlies the mild form [9]. TNSALP replacement therapy is an effective treatment of HPP in infants. The mortality rate of infantile type is 50-100% and patients die due to respiratory infection [10].
We present a case of a 14-month-old girl with the clinical manifestations of rickets, failure to thrive and developmental delay. Hypercalcaemia and low alkaline phosphatase level constitute the main diagnostic laboratory abnormalities if infantile HPP. We aim to share experience.
CASE REPORT
A 14-month-old Saudi girl presented with a 5-day history of cough, fever and shortness of breath. A significant history of recurrent chest infections, hypotonia and abnormal movement was started at three months of age. The patient was born at term via normal vaginal delivery. Parents are healthy, consanguineous and have two other healthy children, the family history had one spontaneous abortion and are of poor socioeconomic status. The patient had a global developmental delay, developmental age around three Months. She vaccinated up to the age of six months. On physical examination, she looks unwell, in respiratory distress, has subtle dysmorphism, unstable vital signs, febrile with temperature 39.2 °C, tachypnea respiratory rate 60 breaths/min, tachycardia, pulse rate 150 beats/min, hypertensive, blood pressure 110/54 mm Hg and an Oxygen saturation of 80% on room air. Her growths showed severe failure-to-thrive. Head exam showed tower-shaped skull (figure: 1) , a wide bulging anterior fontanel and open posterior fontanel, frontal bossing and head lag. Her chest exam showed signs of respiratory distress, bilateral wheezing and crepitation. There were signs of rachitic rosary, Harrison's sulcus and cautery marks (figure: 2). Neurological exam showed microcephaly, generalized hypotonia and hyperreflexia without clonus. Skin was intact and the reminder of the physical examination was unremarkable
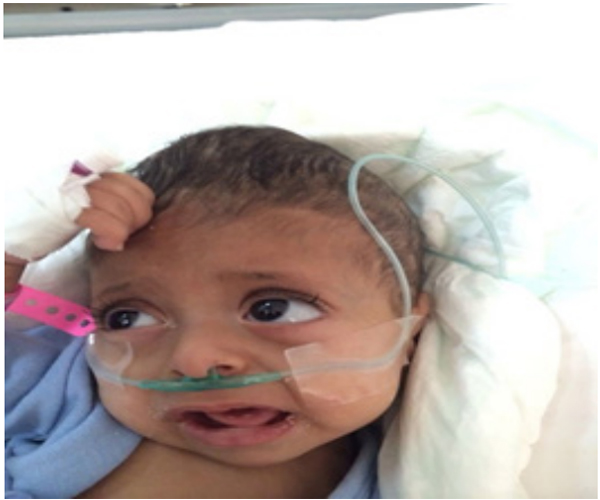
Figure 1: bulge anterior fontanel (cone-shaped) and microcephaly.
The Biochemical workup at presentation showed normal complete blood count (CBC) and Venous blood gasses (VBG). Renal function test & electrolytes showed creatinine 0.4 mg/dl (0.2- 0.4 mg/dl). Urea 10mg/dl (5-18 mg/dl) Sodium 136 mmol/L (130-145 mmol/L) potassium 4.2 mmol/L (4.1-5.3 mmol/L) calcium 11.4 mg/dl (8.8 -10.8 mg/dl) magnesium 2 meq/L (1.7 - 2.1 meq/L) phosphorus 7mg/dL (4-6.5 mg/dL) uric acid 5 mg/dl (1.7 -5.8 mg/dl). Liver function tests was within the normal limit apart from very low level of ALP 14 U/L (150- 420 U/L) .Unremarkable sepsis screening. Hormonal study showed normal Thyroid function test, low 1,25-dihydroxyvitamin D level 6 pg/ml (16- 65 pg/ml) and low parathyroid hormone (PTH) 3 pg/ml (3.6-32 pg/ml).
Electrolyte analysis from urine samples revealed hypercalciuria with high urinary calcium-creatinine ratio 0.95 and nephrocalcinosis by Kidney ultrasound. The Radiographic findings suggest severe osteopenia and rachitic changes. Tandem mass spectrometry (neonatal screening) was unremarkable. Brain magnetic resonance imaging (MRI) was normal apart from Cone-shaped skull and Electroencephalography showed abnormal epileptic discharges.
Patient was admitted to the pediatric intensive care unit (PICU), she received supportive management, empiric antibiotics and antihypertensive medications. She had break through seizures. Although, Anti-epileptic medication was started on her but it was partially controlled after administration of pyridoxine (vitamin B6). Eventually, she developed pulmonary insufficiency, which progressed to life-threatening respiratory failure and death.
Based on clinical, biochemical and radiologic findings; a presumptive diagnosis of infantile HPP was made. Genetic testing not performed.
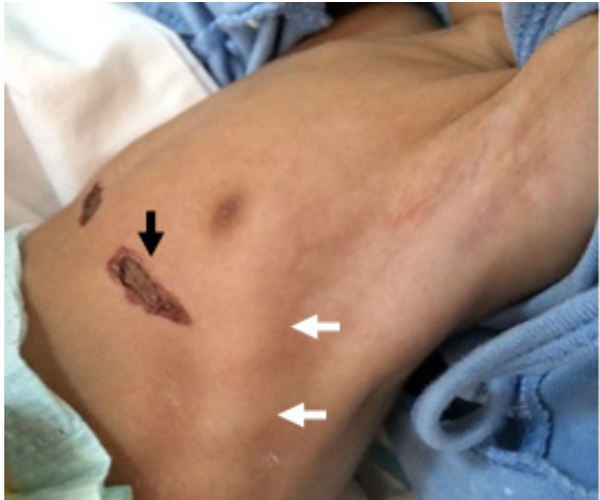
Figure 2: Cautery marks (black arrow) and rachitic rosary (white arrow).
DISCUSSION
Of the cases included in the study, 24 children were male [50%], and 24 were female (50%) in Group I. In Group II, 28 children were male (65.1%), and 15 children were female (34.9%). The mean age was 47.6 - 1.5 months and 72.5 - 1.9 months in Group I and Group II, respectively. The mean body mass index (BMI) was 15.2 - 3.5 kg/m2 and 15.9 - 2.9 kg/m2 in Group I and Group II, respectively. The demographic features and anthropometric measurements of the groups are given in Table 1.
Hypophosphatasia (HPP) was first described by Rathbun (1948) in a 9-week-old male infant [11]. In most cases, it is an Autosomal recessive inheritance, which affects males and females equally [9, 12]. The incidence and prevalence of all forms of HPP are unknown. As this disease resembles rickets, the milder form can go misdiagnosed. It must increase the index of suspicion in children who present with failure-to-thrive, developmental delay, hypotonia and rickets-like signs.
Our case is 14 months old, she was well until the age of three months when she started to have recurrent chest infection, abnormal movements as well as failure-to-thrive. Most of these chest infection attacks required emergency department visit and sometimes admissions. For that reason, she did not complete her vaccination schedule as she was being unwell most of the time.
Later, the child had developmental delay, poor feeding and recurrent choking due to generalized hypotonia which explained by hypercalcemia. Cautery is technique of burning a part of a body, It destroys some tissue in an attempt to remove an undesired growth, or minimize other potential medical harm. Cautery was done many times in the chest because parents do believe in these traditional technique since it is widely practiced in our area (figure: 2). Al-BinAli and his colleagues stated that the perception of parents in favor of practicing cautery for their infants ailments and the main site for cautery was the chest which is due to prolonged cough [13].
On examination, the cranial sutures felt wide with marked bulging of the anterior fontanel, which gives a cone-shaped skull and delayed teething (figure: 1). She also had signs of rachitic rosary (figure: 2).
There was microcephaly, but no craniosynostosis, the premature closure of skull sutures, which considered a feature of the infantile form of HPP. There are sufficient reports of craniocynastosis associated with HPP [12]. As there was no family history to support the diagnosis of hypophosphatasia apart from one abortion and a severe presentation, the most likely mode of inheritance is an autosomal recessive although the genetic study was not done. Alotaibi and her colleague stated that the transmission of the severe type of the disease is mainly by autosomal recessive inheritance, while milder forms may be transmitted as dominant or recessive autosomal traits. [12].
Diagnosis is challenging due to the rareness and variable presentation of symptoms, but Low alkaline phosphatase levels are a signature of HPP [14]. In our patient, serum alkaline phosphatase activity was very low which guide to the diagnosis. Her clinical presentations and investigations supported the diagnosis of infantile hypophosphatasia. Urine test for the presence of phosphoethanolamine was not able to be evaluated. Her convulsions were partially controlled well by pyridoxine. The seizure in most of cases were refractory to conventional antiepileptic drugs but were responsive to pyridoxine [15].
Demirbilek and his colleague described a four-month-old boy, who presented with pyridoxine-responsive seizures in the early neonatal period and was found to have hypercalcemia, skeletal demineralization and increased intracranial pressure [15].
In our case, Hypercalcemia was managed by intravenous fluids. In general, there is some controversy in the literature about the preferred treatment for hypercalcemia in patients with HPP and therapies directed at improving hypercalcemia are expected to worsen bone demineralization [15].
Subsequently, her condition further deteriorated. Although she received maximum attention in the pediatric intensive care unit (PICU), she died from respiratory insufficiency, which is the main cause of death [16].
An established treatment for HPP was not available at the time of presentation of our patient. There was no cure and has had a poor prognosis. In 2015, the U.S Food and Drug Administration (FDA) approved Strensiq (asfotase alfa) as the first approved medical treatment for perinatal, infantile, and juvenile-onset HPP.
Patients of all ages with pediatric-onset HPP are eligible for treatment with this bone-targeted form of TNSALP replacement therapy given by subcutaneous injection. Kosnik-Infinger and his colleagues did a study, which reveals enzyme replacement therapy (ERT) administered either pre- or postoperatively paired with cranial vault remodeling strategies can yield improved neurological outcomes in children affected by HPP [17].
Transplantation Therapy by Using Bone Fragments and Cultured Osteoblasts was tried in infantile type and it can improve mineralization [18]
However, multidisciplinary care remains the core of treatment of infantile hypophosphatasia encompassing nutritional support, adjustment of calcium and phosphate intake, monitoring of vitamin D levels, careful and personalized physical therapy, and regular dental monitoring and care [18].
CONCLUSION
Hypophosphatasia should be highly suspected when serum ALP is found to be low. It is a rare disease and has a variety of presentations. Our case was having a classical presentation of infantile type and died from chest infection. Early diagnosis of such cases is essential for subsequent management and achieving a good outcome.
REFERENCES
- Hu CC, King DL, Thomas HF and Simmer JP. (1996). A clinical and research protocol for characterizing patients with hypophosphatasia. Pediatr Dent. 18(1): 17-23.
- Deeb A, Bruce S, Morris A and Cheetham T. (2000). Infantile hypophosphatasia: disappointing results of treatment. Acta Paediatr. 89(6): 730-733
- Rauch F and Glorieux FH. (2004). Osteogenesis imperfecta. Lancet. 363(9418): 1377-85.
- Michael P and Whyte. (1994). Hypophosphatasia and the Role of Alkaline Phosphatase in Skeletal Mineralization. Endocrine Reviews. 15(4): 439-461
- McMillan JA, Feigin RD, DeAngelis C and Jones J. (2006). Oskis Pediatrics. 4th ed.
- Whyte MP, Teitelbaum SL, Murphy WA, Bergfeld MA, et al. (1979). Adult hypophosphatasia. Clinical, laboratory, and genetic investigation of a large kindred with review of the literature. Medicine (Baltimore). 58(5): 329-47.
- Fallon MD, Teitelbaum SL, Weinstein RS and Goldfischer S, et al. (1984). Hypophosphatasia: clinicopathologic comparison of the infantile, childhood, and adult forms. Medicine (Baltimore). 63(1): 12-24.
- Seshia SS, Derbyshire G, Haworth JC and Hoogstraten J. ( 1990). Myopathy with hypophosphatasia. Arch Dis Child. 65(1): 130-131.
- Mornet E and Nunes ME. (2007). Hypophosphatasia. Gene Reviews.
- Whyte MP, Rockman-Greenberg C, Ozono K and Riese R, et al. (2016). Asfotase Alfa Treatment Improves Survival for Perinatal and Infantile Hypophosphatasia. J Clin Endocrinol Metab. 101(1): 334-42.
- Rathbun J. (1948). Hypophosphatasia. Am. J. Dis. Child. 75: 822-831, [PubMed: 18110134, related citations.
- Maha Alotaibi and Al Qassmi Amal. (2017). New homozygous mutation in ALPL gene in Saudi patient with infantile hypophosphatasia. Open acses test.
- Ali M. Al-Binali, Mohammed A. Al-Huneif, Safa M. AlHaider and Ossama M. (2014). Perception of Cautery Healing Effect among Infants Parents at the Southwestern Area of Saudi Arabia. Global Journal of Medical Research. Global Journals Inc. 14.
- Agns Bloch-Zupan. (2016). Hypophosphatasia: diagnosis and clinical signs a dental surgeon perspective. International journal of pediatric dentistry. 26(6): 426-438.
- Huseyin Demirbilek, Yasemin Alanay, Ayfer Alikasifoglu and Kandemir N. (2012). Hypophosphatasia Presenting with Pyridoxine-Responsive Seizures, Hypercalcemia, and Pseudotumor Cerebri: Case Report. lin Res Pediatr Endocrinol. Mar. 4(1): 34-38.
- Etienne Mornet (2007). Hypophosphatasia. Orphanet Journal of Rare. 2:40 doi.org/10.1186/1750-1172-2-40
- Libby Kosnik-Infinger, Craig Gendron, Christopher B. Gordon and a Brian S. Pan. et al. ( 2015). Enzyme replacement therapy for congenital hypophosphatasia allows for surgical treatment of related complex craniosynostosis: a case series. Neurosurg Focus 38 (5): E10.
- Cahill RA, Wenkert D, Perlman SA, Steele A, et al. (2007). Infantile Hypophosphatasia: Transplantation Therapy Trial Using Bone Fragments and Cultured Osteoblasts. The Journal of Clinical Endocrinology & Metabolism. 92(8): 2923-2930.