Previous Issues Volume 3, Issue 2 - 2019
Everolimus for Treatment Tuberous Sclerosis Complex with Refractory Epilepsy: Management and Out Comes. A Case Report
Nikolas Andre Mata-Machado*, Anna Carolina Paiva Costa T Figueiredo
Corresponding author: Nikolas Andre Mata-Machado, Department of Pediatric Neurology, University of Illinois, 1801 west Taylor Street, Suite 2E (MC 686), Chicago, IL, USA. Email: [email protected]
Received Date: Nov 21, 2019 Published Date: Dec 26, 2019 Copyright © 2019 Mata-Machado NA.
Citation: Mata-Machado NA. (2019). Everolimus for Treatment Tuberous Sclerosis Complex with Refractory Epilepsy: Management and Out Comes. A Case Report. Mathews J Cytol Histol 3(2): 13.
ABSTRACT
Tuberous sclerosis complex (TSC) is an autosomal-dominant neurocutaneous disorder characterized by multiple hamartomas and neurodevelopmental disorders. Often refractory, seizures are considered the primary cause of neurologic morbidity. Patients with TSC present Mutations of the TSC1 and TSC2 genes, in TSC patients, activates the mTOR pathway. Reduction in tumor size (SEGAs, renal angiomyolipoma) and epilepsy, lungs function and skin features (especially facial angiofibroma) has been evaluated as a benefit of mTOR inhibitors treatment. We report a case of a 5 year old girl with the features of TSC like hypomelanotic macules, facial angiofibromas, shagreen patch, refractory epilepsy and angiolipomas of the kidney, in who we started treatment with everolimus.
Keywords: mTOR Inhibitors; Epilepsy; Everolimus; Seizures; Tuberous Sclerosis complex.
INTRODUCTION
TSC is an autosomal dominant disorder that variably affects the CNS, skin, kidneys, heart, and other organs. Birth incidence is estimated to be 1:5800 [1]. Inactivating mutations in TSC1 gene on chromosome 9q34 [1,2], or TSC2 gene on chromosome 16p13.3[3], which encodes for the proteins hamartin and tuberin, respectively, are considered the primarily caused of TSC [2]. These mutations subsequently lead to hyperactivity of the mTOR pathway [3]. Most patients with TSC are born with at least one detectable mutation and are thus Heterozygous for either TSC1 or TSC2, most TSC patients have inactivating mutations of the other functional allele (loss of heterozygosity) [4] which is detectable within peripheral and brain lesions [5]. Other putative mechanisms leading to TSC1 or TSC2 may include haploinsuffiency but need further investigations [6].
Hypomelanotic macules, shagreen patch, ungual fibromas, and facial angiofibromas are some of the TSC skin features. They are usually present inover 90% of individuals with TSC, although none is pathognomonic [6]. Renal angiomyolipomas, histologically benign tumors with varying amounts of vascular tissue, fat, and smooth muscle, appear in 75 to 80% of patients older than 10 years of age with TSC [7].
Cortical hamartomas (“tubers”), subependymal nodules, subependymal giant cell astrocytomas (SEGAs), white matter abnormalities, and microscopic abnormalities such as microdygenesis, heterotopic gray matter and lamination defects are brain lesions often observed in TSC. Transmantle cortical dysplasia, hemimegalencephaly [8-10] and schizencephaly are rarely seen [11].
Intellectual developmental disorders, epileptic seizures and behavioral abnormalities are some of the main neurological manifestations, although there are diagnosed patients with little or no neurologic impairment [12,13]. The mechanisms thought to be responsible for the neurological lesions are impaired neuronal migration along radial glial fibers and also anomalous proliferation of glial elements.
Seizures in different types occur in 80 to 90% of TSC patients. Commonly, TSC patients present with intellectual developmental disorder associated with epilepsy, although mots of individuals have epilepsy but normal intellectual development. There is no correlation between the number of subependymal lesions and clinical severity of TSC. Severe cognitive impairment and difficult to control seizure are usually observed in patients with numerous cerebral cortical lesions shown by MRI [14,15]. Long-term cognitive impairment in patients with more cortical MRI lesions are prone to be exhibited in infantile spasms.
Intended to serve as a comprehensive framework for evaluating and treating patients with TSC, in 2012, the International Tuberous Sclerosis Complex Consensus Conference updated previous guidelines [16] shown below (Table 1).
Table 1: Diagnostic Criteria for Tuberous Sclerosis Complex.
|
Genetic diagnostic criteria |
The identification of a TSC1 or TSC2 pathogenic mutation in DNA from normal tissue is sufficient to make a definitive diagnostic of TSC |
|
|
Clinical diagnostic criteria |
Major features |
Facial angiofibromas or forehead plaque Non-traumatic ungual or periungual fibroma Hypomelanotic macules (> three) Shagreen patch (connective tissue nevus) Cortical tuber Subependymal nodule Subependymal giant cell astrocytoma(SEGA) Multiple retinal nodular hamartomas Cardiac rhabdomyoma, single or multiple Lymphangiomyomatosis Renal angiomyolipoma |
|
Minor features |
Multiple randomly distributed pits in dental enamel Hamartomatous rectal polyps Bone cysts Cerebral white-matter "migration tracts" Gingival fibromas Non renal hamartoma Retinal achromic patch "Confetti" skin lesions Multiple renal cysts |
|
The presence of two major features or one major feature plus at least two minor features is considered sufficient for the diagnosis of TSC. The presence of one major feature alone or two minor features is considered possible diagnosis.
In this report, we describe the case of a patient with TSC in whom TSC-related symptoms improved after treatment with the mTOR inhibitor everolimus.
CASE REPORT
A 5 year old girl reported to the neurology department with the diagnostic of TSC and intractable epilepsy who came to establish care. Parents gave a medical history of a full term neonate with a fetal US findings concerning of TSC. She was found to have tumors in her heart in the first week of life work up. The seizures onset was after her second month immunization and had progressed in intensity and severity even with anti-seizure medication treatment, including Vagabatrin. When care was started the patient was having a range from 3 to 10 daily seizures. Was also observed an intellectual and eurodevelopment delay. She has been spoon fed and does not have a G-tube. No family history was found.
Patient examination revealed multiple papular lesions of light brown coloured of about 5 -10 mm size, seen on nasal bridge and extending to the cheek region usually in a butterfly like distribution suggestive of facial angiofibromas (Figure 3). Hypomelanotic white macules (ash leaf macule) seen on the chest and right posterior ankle (Figure 1). Thick leathery skin area that is dimple like an orange peel seen on right flank side of the patient suggestive of shagreen patches (Figure 2). Neurologically impaired: non-verbal, wheel-chair dependent, no cranial nerves deficits, dystonia present in bilateral upper and lower extremities.
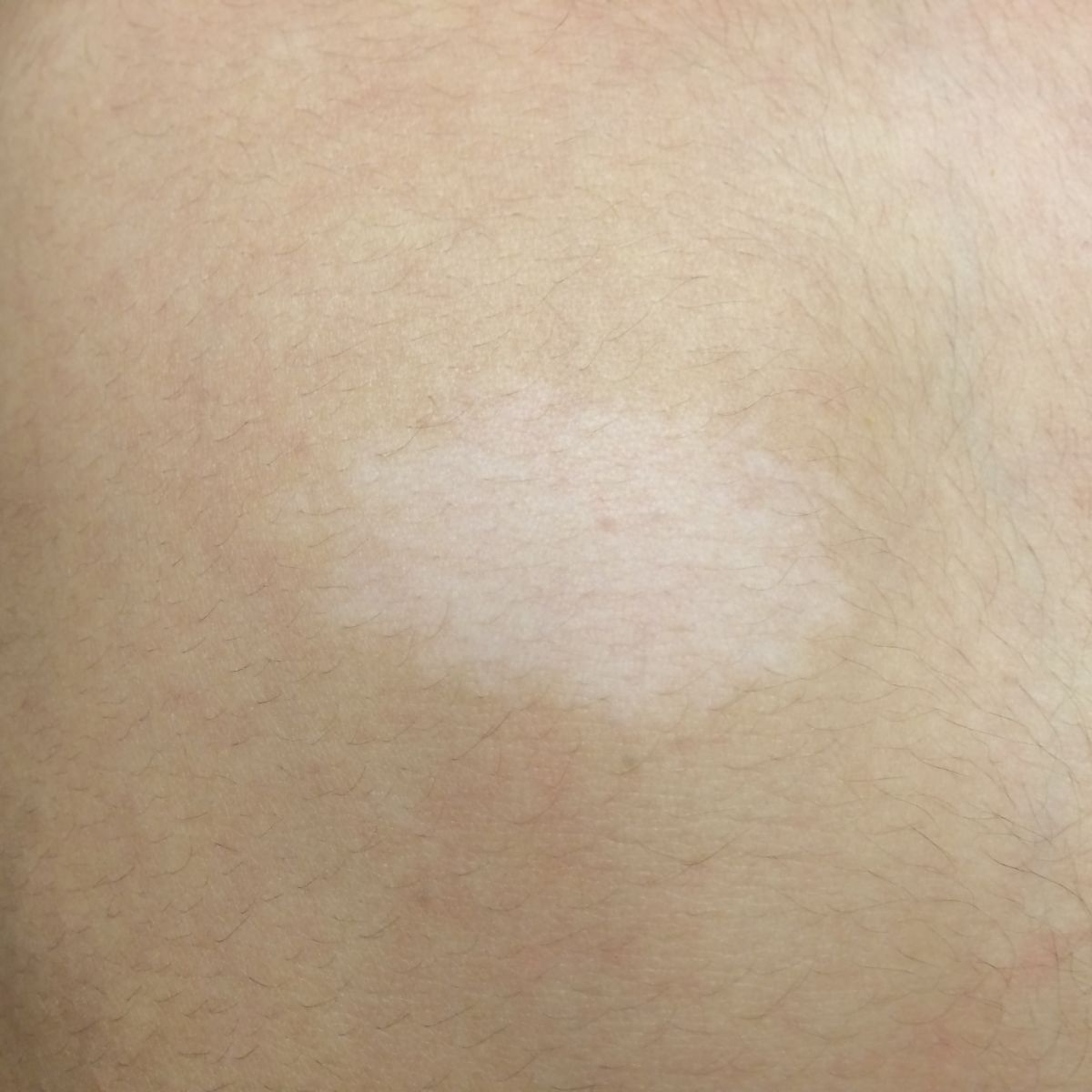
Figure 1: Hypomelanotic macule (ash leaf macule) over the anterior thoracic region.
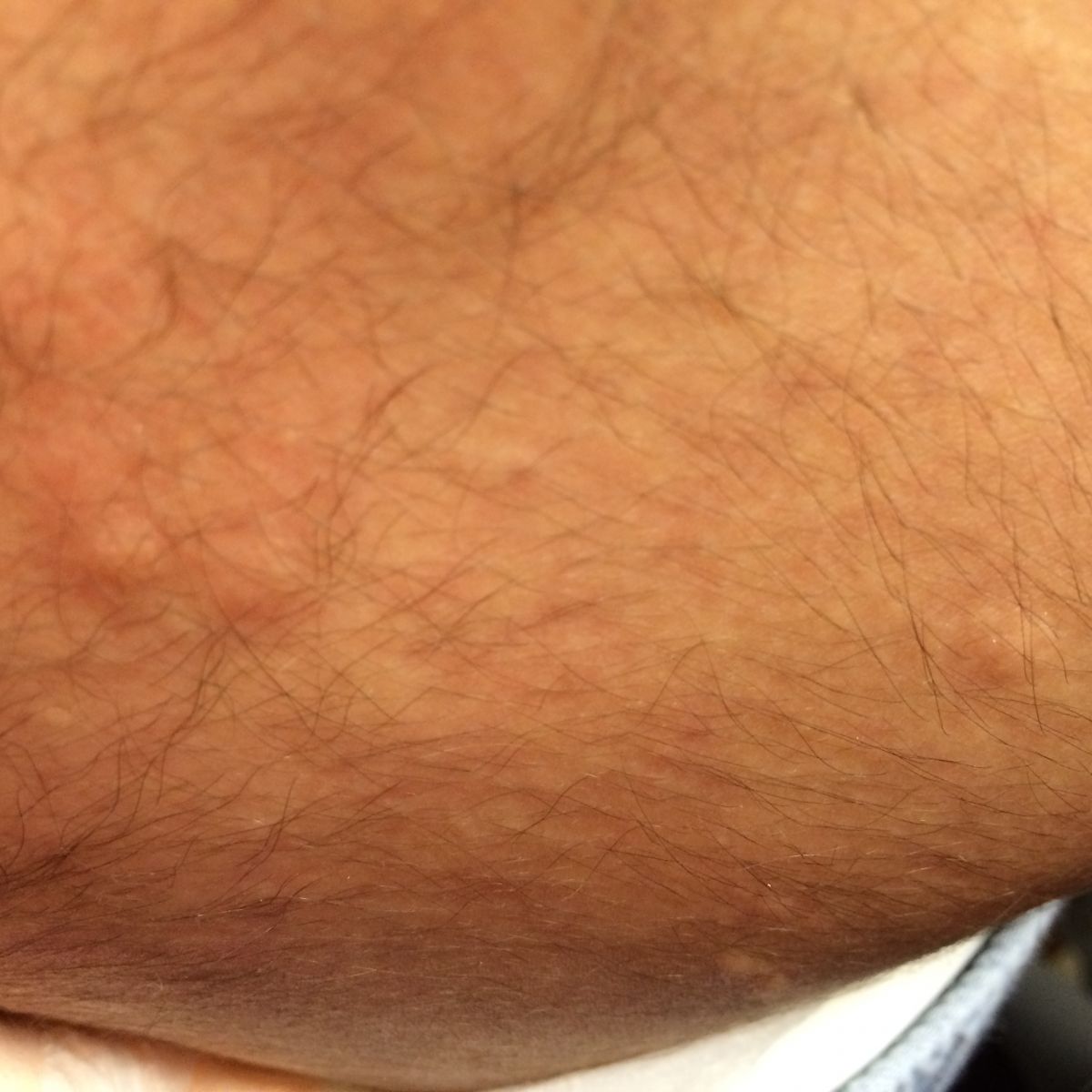
Figure 2: Single shagreen patch over the lumbosacral region.
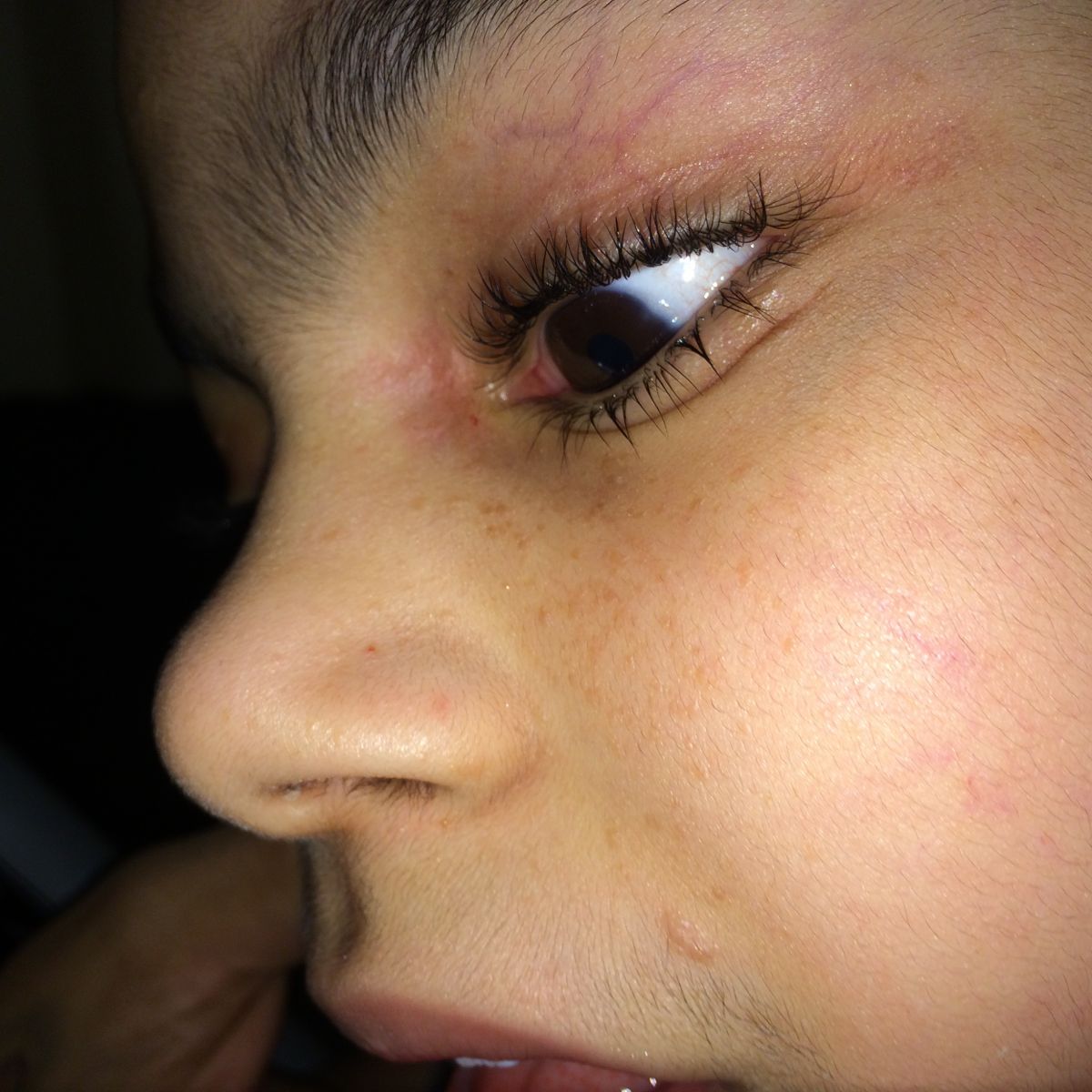
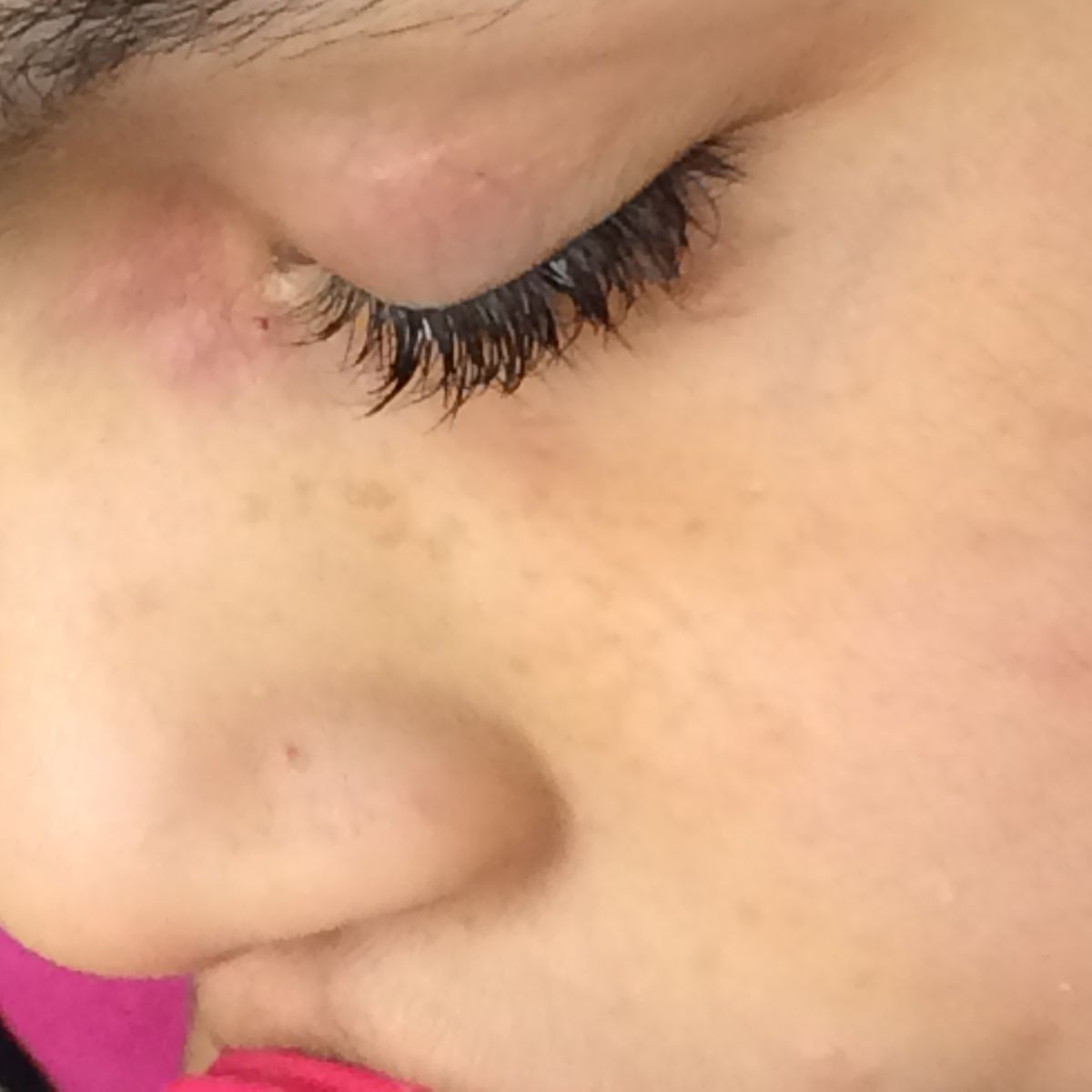
Figure 3: Wart-like, waxy lumps consisting of angiomatous and fibrous tissue (angiofibromas) over the nasolabial fold and alar nose.
A prolonged EEG showed a generalized pattern consistent with Lennox-Gastuat syndrome. Renal US showed innumerable angiolipomas bilateral in the kidneys. Head CT demonstrated sub-ependymal nodules as well as subcortical tubers, some with dystrophic calcifications.
Since the care was established has been challengeable to control intensity and severity of seizures. There is a list of medications that was tried and failed as known as VPA, Topiramate, Zonegran, Felbamate, ACTH, Vigabatrin, Ketogenic diet and VNS(do not work in high parameters, currently on minimal parameters). In a first approach we add lamictal in a progressive way, stood with clobazam, rufinamide and we consider to enroll her in everolimus trial. After nephrologist, cardiologist and ophthalmologist evaluation we started work up preparation for everolimus use. Was ordered lung function test, BUN, creatinine, ALT,AST, CBC, Lipid profile, glucose level, HbA1,CMP, UA, urine culture, chest X-ray, brain MRI, Neuropsychological testing and Everolimus level in 2 weeks from start. Except for Brain MRI that showed 2 small SEGAs over the foramen of Monro; no other alterations was found and the drug was initiated at the dose of 3mg.
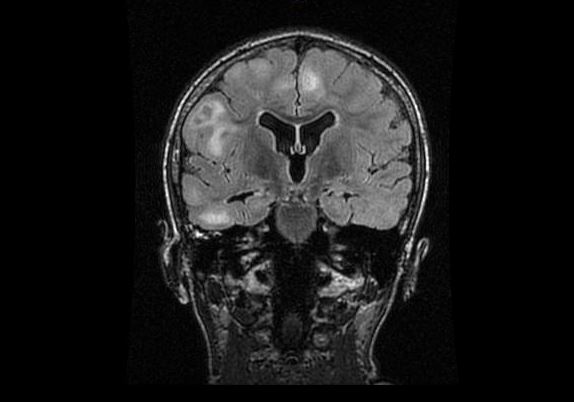
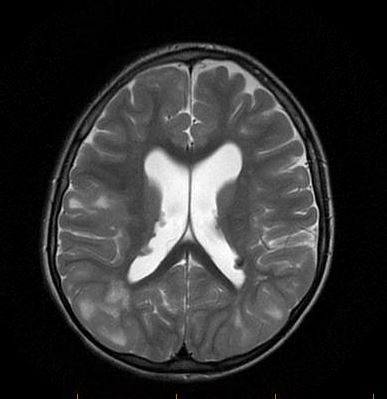
MRI demonstrate numerous subcortical hamartomas
MRI demonstrate subependymal nodules near the foramina of Monroe bilaterally
After 4 weeks of use she had an improvement on the facial angiofibromas, she became more social and had less spasticity over the upper and lower extremities, but her alkaline phosphatase went up 20 times and we had to stop it. At the second attempt of everolimus, her scalp skin was pilling off and on the third attempt she had bumps on her scalp that was showed by pathology be due to Staphylococcus. Therefor, the drug was stopped again and cephalexin was started and continued after the patient was cleared in a prophylaxis base to re-start everolimus. Once more we re-started the drug and for this time on the patient is having a good response with the medication without any major concerns We had a follow up brain MRI that showed multiple subependymal nodules with no change in size and number, and also a abdominal MRI which showed more than 100 angiomyolipomas(biggest around 1,2x1.4cm) . The patient is a close follow up with her nephrologist. During this time she still had refractory seizures with periods of worsening symptoms, but seemed to be in a better control (seizures every other day) with maximized dose of lamictal, Onfi and Banzel.
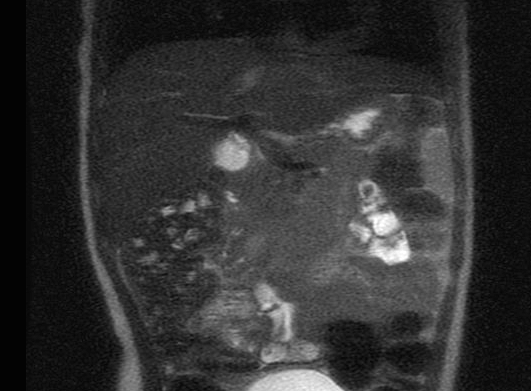
MRI demonstrate bilateral angiomyolipomas in the kidneys.
DISCUSSION
Trough the intermediary signaling protein Rheb (Ras Homolog Enriched in Brain) TSC1 and TSC2 form a complex that regulates the mammalian target of rapamycin complex 1 (mTORC1) [17]. Renal angiomyolipomata [18,19] SEGAs, [20] and lymphangioleiomyomatosis (LAM) are some of the TSC manifestations that benefited from pharmacologic inhibitors of mTORC1, specially sirolimus and everolimus. Seizures improvement was also reported by case reports and preclinical studies [21-25].
Treatment with conventional antiepileptic medications (AEDs) may lead to successful seizure control in some TSC patients, although in of them, seizure control was not obtained with available medical and surgical therapies. In those refractory epilepsy, there is a high prevalence of cognitive impairment and neuropsychiatric and developmental disorders.
The specific mechanism(s) responsible for the positive changes is not known. Traditional AEDs, which target ion channels, synaptic receptor function, or neurotransmitter release directly are thought to act differently from mTORC inhibitors. Everolimus, through the inhibition of mTORC1 has shown to act by an indirect mechanism including protein turnover and synthesis, which may influences synaptic excitability, longterm depression and potentiation, and neuronal plasticity.
CONCLUSION
In this report, we described the case of a patient who presented with intractable epilepsy, intellectual and neurodevelopmental delay, bilateral angiomyolipomas in the kidneys, and cutaneous manifestations of TSC. In summary, after multiple attempts of everolimus treatment that had to be withdraw because of side effects, the patient has had beneficial effects on this drug with considerable improvement on the cutaneous TSC features, reduced seizure frequency and duration, which was previously refractory to currently available AEDs. Better behavior and quality of life has been related with this improvement in seizure control. Treatment-related side effects were limited, providing a favorable risk: benefit ratio.
REFERENCES
- O’Callaghan F, Shiell A, Osborne J and Martyn CN. (1998). Prevalence of tuberous sclerosis estimated by capture-recapture analysis. Lancet. 352 (9114): 318–319.
- European Chromosome 16 Tuberous Sclerosis Consortium (1993) Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75(7): 1305–1315.
- Kwiatkowski DJ. Rhebbing up mTOR. (2003). new insights on TSC1 and TSC2, and the pathogenesis of tuberous sclerosis. Cancer Biology and Therapy. a;2(5): 471–476.
- Green AJ, Smith M and Yates JR. (1994). Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nature Genetics. 6(2): 193–196.
- Kwiatkowski DJ and Manning BD. (2005). Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Human Molecular Genetics.14: R251–R258.
- Roach ES, Gomez MR and Northrup H. (1998). Tuberous sclerosis complex consensus conference: Revised clinical diagnostic criteria. J Child Neurol. 13(12): 624-624.
- Ewalt DE, Sheffield E and Sparagana SP. (1998). Renal lesion growth in children with tuberous sclerosis complex. J Urol. 160(1): 141- 141.
- Galluzi P, Cerase A and Strambi M. (2002). Hemimegalencephaly in tuberous sclerosis complex. J Child Neurol. 17(9): 677-680.
- Parmar H, Patkar D, Shah J and Patankar T. (2003). Hemimegalencephaly with tuberous sclerosis : a longitudinal imaging study. Australas Radiol. 47(4): 438-442.
- Sakuma H, Iwata O and Sasaki M. (2005). Longitudinal MR findings in a patient with hemimegalencephaly associated with tuberous sclerosis. Brain Dev. 27(6): 458-461.
- Huntsman RJ, Sinclair DB and Richer LP. (2006). Tuberous sclerosis with open lipped schizencephaly. Pediatr Neurol. 34(3): 231-234.
- Gomez MR, Sampson JR and Whittemore VH (Eds). (1999). Tuberous Sclerosis Complex. [3rd Ed.], Oxford University Press, New York, USA.
- Curatolo P (Ed). (2003). Tuberous Sclerosis Complex: From Basic Science to Clinical Phenotypes. Mac Keith Press, London, UK.
- Roach ES, Williams DP and Laster DW. (1987). Magnetic resonance imaging in tuberous sclerosis. Arch Neurol. 44: 301-303.
- Goodman M, Lamm SH, Engel A, Shepherd CW, et al. (1997). Cortical tuber count: A biomarker indicating cerebral severity of tuberous sclerosis complex. J Child Neurol. 12(2): 85-90.
- Northrup H and Krueger DA. (2013). International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 49(4): 243-254.
- Huang J, Manning B. (2008). The TSC1-TSC2 complex: a molecular switchboard controlling cellgrowth. Biochem J. 412(2): 179–190.
- Bissler J, McCormack F, Young L, Elwing JM, et al. (2008). Sirolimus for angiomyolipomata in tuberous sclerosis or lymphangioleiomyomatosis. N Engl J Med. 358(2): 140–151.
- Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA et al. (2013). Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 381(9869): 817–824.
- Franz DN, Leonard J, Tudor C, Chuck G, et al. (2006). Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 59(3): 490–498.
- Meikle L, Pollizzi K, Egnor A, Kramvis I, et al. (2008). Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 28(21): 5422–5432.
- Meikle L, Talos D, Onda H, Pollizzi K, et al. (2007). A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 27(21): 5546–5558.
- Muncy J, IJ B and Koenig M. (2009). Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol. 24(4): 477.
- Zeng LH, Rensing NR and Wong M. (2009). The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 29(21): 6964–6972.
- Zeng LH, Xu L, Gutmann DH and Wong M. (2008). Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 63(4): 444–453.