Previous Issues Volume 3, Issue 1 - 2019
Acquired C1-Esterase Inhibitor Deficiency Associated with Burkitt Lymphoma
John G Prichard1*, Delilah D Coconubo2, Melanie Gero3, Behzad Noorbehesht4, Michelle Azimov5
Corresponding Author:John G Prichard* Medical Director, Immunology Clinic, Ventura County Medical Center, California, USA.
1Department of Medicine, Immunology Clinic,Ventura County Medical Center, USA 2Immunology Clinic, Ventura County Medical Center 300 Hillmont, Bld 340, Ventura, USA 3Department of Pathology, Ventura County Medical Center, 3291 Loma Vista Road, Ventura, USA 4Palms Imaging Center, 1901 Outlet Center Drive, Suite 120, Oxnard, USA 5Department of Hematology and Oncology, VenturaCounty Medical Center, 300 Hillmont, Bld 340, Ventura, USA
Received Date: Aug 26, 2019 Published Date: Sep 16, 2019
Copyright © 2019 John G Prichard
Citation: John G Prichard, Delilah D Coconubo, Melanie Gero, Behzad Noorbehesht, et al. (2019). Acquired C1-Esterase Inhibitor Deficiency Associated with Burkitt Lymphoma. M J Immu. 2(2): 008.
Abstract
C1-esterase inhibitor deficiency results in recurrent episodes of angioedema. Acquired deficiency has been associated with B-cell lymphoproliferative disorders, rheumatologic diseases or, in a small proportion of patients, the cause remains unknown. Of the malignancies associated with acquired C1-esterase deficiency, indolent lymphomas and plasma cell disorders are the most common. Aggressive lymphomas are rarely reported in such cases. We report the association of Burkitt lymphoma, a highly aggressive B-cell malignancy, which evolved seven months after the initial symptoms of acquired angioedema in a fifty-five year old man. Following treatment of the lymphoma, episodes of angioedema ceased. An association of acquired angioedema with Burkitt lymphoma has not previously been reported.
Keywords: Acquired angioedema, C1-Esterase inhibitor deficiency, Aggressive B-cell lymphoma, Burkitt lymphoma
Introduction
Angioedema is characterized by the abrupt onset of vascular permeability resulting in episodic, localized edema of soft tissues of the upper airway, skin or gastrointestinal tract. It is due to functional deficiency of C1-esterase. Such a deficiency may be hereditary or acquired. The acquired form, usually occurring in older adults, is associated with autoimmune or lymphoproliferative disorders. In the latter instance, both plasma cell disorders and indolent lymphomas predominate. Aggressive lymphomas are rarely associated with acquired C1-esterase deficiency. We report the unusual occurrence of acquired angioedema preceding the diagnosis of Burkitt lymphoma, a highly aggressive B-cell malignancy. This association has not been reported previously, and illustrates the importance of continued observation despite normal studies at the time of diagnosis of C1-esterase deficiency.
Case Report
A fifty-five year old man was admitted to hospital because of edema involving his lip, tongue and uvula. He required emergency intubation. His past medical history included rheumatoid arthritis diagnosed eight years earlier, type II diabetes mellitus, and hypertension. There was no family history of angioedema.
Three months before admission angioedema involving his lip occurred and was associated with non-pruritic erythematous plaques. An angiotensin converting enzyme inhibitor, taken for treatment of hypertension, was discontinued. Two months before admission facial and lip edema occurred and resolved with non-specific treatment. That episode was soon followed by hemifacial edema and transient, generalized abdominal pain. Two weeks before admission an episode of scrotal edema occurred and resolved. CT scan of the abdomen and pelvis showed no intraabdominal or pelvic adenopathy. CH50 was <10 (N=31-60), a functional assay for C1 inhibitor was 36% (N= >68%). C1q was undetectable. A diagnosis of acquired C1- esterase deficiency was made.
His hospital course was complicated by hospital acquired pneumonia. He was treated with fresh frozen plasma (human C1-esterase inhibitor therapy was unavailable) and extubated on the fifth hospital day. Subsequent laboratory studies disclosed a normal complete blood count and serum lactate dehydrogenase (LDH). Serum protein electrophoresis was normal. Repeat CT scan of the abdomen showed no new abnormalities. He was placed on danazol and human C1-esterase inhibitor was made available. No further episodes of angioedema occurred.
Seven months following the first episode of angioedema, he developed severe and progressive pain in his left thigh and inguinal region. His left leg was massively swollen and tender. Left inguinal adenopathy was present. The LDH was 725 IU/L, three times the upper limit of normal. Serology was negative for human immunodeficiency virus (HIV). The complete blood count was normal and free of circulating malignant cells. Magnetic resonance imaging (MRI) of the left lower extremity showed a bone marrow centric lesion involving most of the distal femoral diaphysis and surrounding soft tissue. Additional bony lesions were seen in the pelvis and both femoral heads. Bulky adenopathy was present in the popliteal, inguinal and external iliac regions. Positron emission tomography (PET-CT) (Figure 1a and 1b) showed extensive disease above and below the diaphragm. No abnormal uptake was seen in the liver or spleen. Brain MRI showed a 20 mm × 28 mm enhancing extra-axial mass arising from the left sphenoid wing. Analysis of cerebral spinal fluid was negative for tumor cells by flow cytometry.
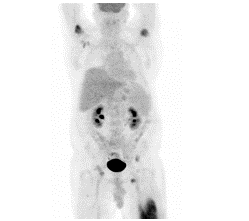
Figure 1A: Fig1a Baseline PET/CT: Maximum Intensity Projection image shows a large left thigh lesion measuring 18 cm in length. Maximal SUV is 9.2. There are additional smaller lesions in the bilateral humeral heads, right femoral neck, and bilateral pelvic bones. There is also mild right axillary and bilateral pelvic lymphadenopathy.
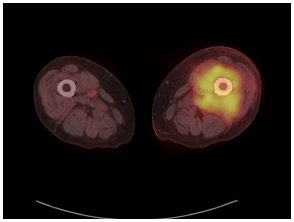
Figure 1B: Baseline PET/CT: Axial fused image at the level of the midthighs demonstrates a large moderately hyper-metabolic infiltrative soft tissue mass in the left thigh surrounding the femur and involving the anterior and posterior musculature and the bone marrow.
Biopsy of an inguinal lymph node showed a fairly uniform population of medium sized round cells with numerous apoptotic cells, imparting a starry sky appearance (Figure 2a). Immuno-histochemical staining of the tumor cells showed strong positive staining with CD10, CD20, and BCL6. Staining for Ki67 (Figure 2b) and MYC were diffusely positive with staining approaching 100%. Stains for BCL2, EBER, TdT, CD30 and CD34 were negative. Fluorescent in situ hybridization (FISH) showed MYC (8q24) rearrangement without detection of MYC amplification. There was no evidence of BCL2 or BCL6 rearrangement, excluding a double or triple hit diffuse large B-cell lymphoma. The findings were most consistent with Burkitt lymphoma.
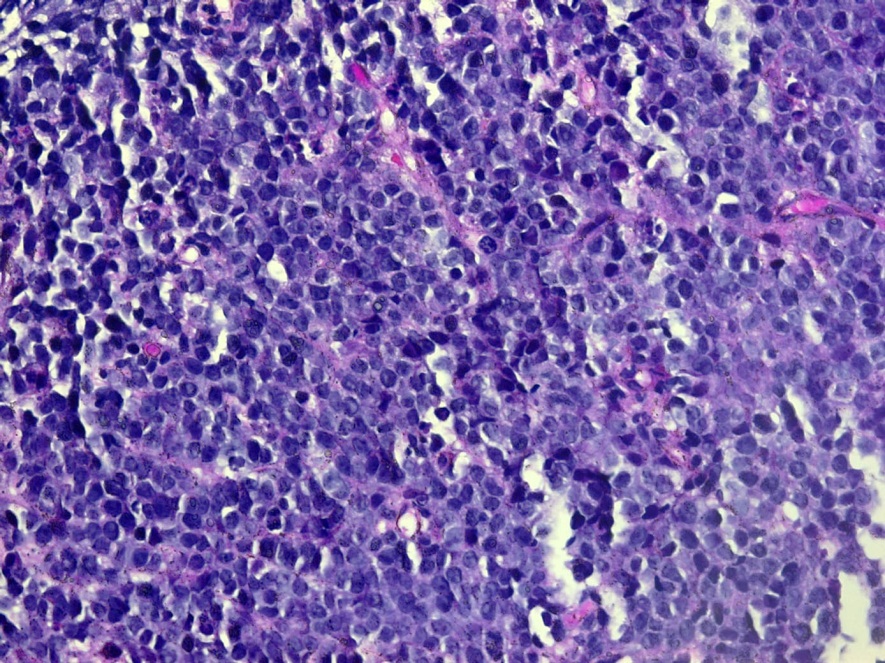
Figure 2A: (H&E stain 400x) Burkitt Lymphoma consisting of a diffuse, uniform population of intermediate-sized cells with a starry sky appearance. There are mitotic figures and numerous tingible body macrophages containing apoptotic debris.
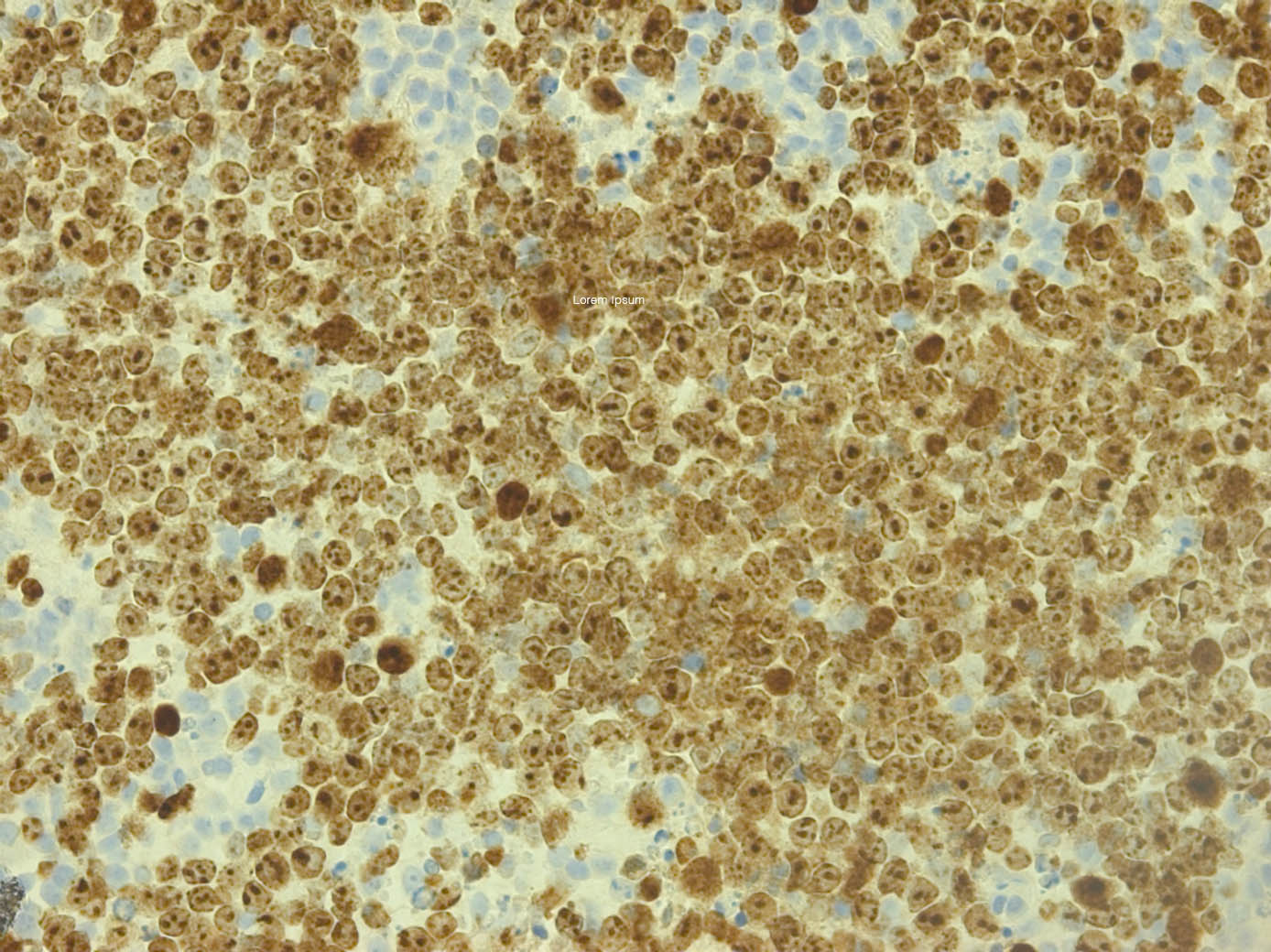
Figure 2B: (Ki-67 stain 200x) The viable Burkitt lymphoma cells have a Ki67 positive proliferation rate of nearly 100%.
He was begun on Rituxan, etoposide, prednisone, vincristine, doxorubicin and cyclophosphamide (R-EPOCH) for six cycles followed by two cycles of intravenous methotrexate, 8000 mg. Post treatment, a brain MRI showed no evidence of residual disease. At ten months following his last treatment, there had been no recurrence of angioedema and no evidence of recurrent lymphoma. Following discontinuation of danazol, CH50, C1-esterase inhibitor function, and C1-esterase inhibitor protein values were normal.
Discussion
Both hereditary and acquired C1-esterase inhibitor deficiency results in episodes of angioedema, which can cause sudden airway obstruction [1]. The acquired form occurs in adults who lack a family history of angioedema. The diagnosis is confirmed by very low or undetectable C1-esterase protein, C1-esterase activity <40% and low or absent total hemolytic complement (CH50). The pathogenesis of acquired angioedema is not precisely understood, but current evidence suggests a process whereby C1-esterase inhibitor is functionally inhibited by autoantibodies [2]. Thus, acquired angioedema has been associated with autoimmune disorders in roughly 10% of cases [3]. In an additional 40% of cases, monoclonal gammopathy has been documented [4]. The remaining patients are found to have an underlying malignancy, most often an indolent non-Hodgkin B-cell lymphoma. Of the non-aggressive lymphomas, marginal zone lymphoma is most often encountered, but chronic lymphocytic leukemia and mantle cell lymphoma have also been reported [5]. Marginal zone lymphomas, both splenic and nodal forms, are associated with autoimmune phenomena [6].
Burkitt lymphoma is a highly aggressive B-cell lymphoma. The most common type in the United States and Western Europe is the sporadic form, which is not associated with HIV, and is most often seen in children. In adults, however, Burkitt lymphoma represents less than 1% of all non-Hodgkin lymphomas. It is considered the fastest growing malignancy in humans [7] and frequently presents at extranodal sites [8]. Burkitt lymphoma is considered a mature B-cell neoplasm of germinal center origin [9] and thus expresses CD10, BCL6, PAX5, CD19, and CD20. Membrane IgM is uniformly present. Abnormalities of chromosome 8, usually a translocation with chromosome 14, allow the constitutive expression of the MYC gene, a transcriptional activator [10], which accounts for the tumor’s high replication capacity, indicated by Ki67 positivity approaching 100%.
In the case presented here, Burkitt lymphoma did not become evident until seven months following the episode of angioedema involving his upper airway. Severe leg pain and swelling was of fairly sudden onset, and progressed very rapidly. His initial evaluation at that time disclosed stage IV disease, including metastasis adjacent to the brain. A delay between onset of angioedema and discovery of a lymphoproliferative disorder is not unusual. Indeed, such a disorder may not become evident for several years following diagnosis of C1-esterase deficiency [5], reflecting the indolent nature of most of the associated diseases. Discovery of an indolent lymphoproliferative disorder usually does not represent an oncologic emergency. Burkitt lymphoma, on the other hand, is extremely aggressive and warrants urgent staging and treatment. This case illustrates the need for prompt investigation of new complaints, or laboratory abnormalities, during monitoring of patients with acquired C1-esterase deficiency where the cause is not known.
References
- Agustoni A and Cicardi M. (1992). Hereditary and acquired C1-inhibitor deficiency: Biological and clinical characteristics in 235 patients. Medicine. 71(4): 206-215.
- Jackson J, Sim RB, Welan A and Feighery C. (1986). An IgG autoantibody which inactivates C1-inhibitor. Nature. 323(6090): 722-724.
- Zingale LC, Castelli R, Zanichelli A and Cicardi M. (2006). Acquired deficiency of the inhibitor of the first complement component: Presentation, diagnosis, course and conventional management. Immunol Allergy Clinic. 26(4): 669-690.
- Gobert D, Paule R, Ponard D, Levy P, Frémeaux-Bacchi V, et al. (2016). A nationwide study of acquired C1-inhibitor deficiency in France: Characteristics and treatment responses in 92 patients. Medicine. 95(33): e4363.
- Castelli R, Deliliers DL, Zingale LC, Pogliani EM and Cicardi M. (2007). Lymphoproliferative disease and acquired C1-inhibitor deficiency. Haematologica. 92(5): 716-718.
- Piris MA, Onaindia A and Mollejo M. (2017). Splenic marginal zone lymphoma. Best Pract Res Clin Haematol. 30(1-2): 56-64.
- O’Malley DP, Auerbach A and Weiss LM. (2015). Practical applications in immunohistochemistry: Evaluation of diffuse large B-cell lymphomas. Arch Pathol Lab Med. 139(9): 1094-1107.
- Kalisz K, Alessandrino F, Beck R, et al. (2019). An update on Burkitt lymphoma: a review of pathogenesis and multimodality imaging assessment of disease presentation, treatment response, and recurrence. Insights into Imaging. 10(1): 56.
- Swerdlow SH, Campo E and Pileri SA. (2016). The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 127(20): 2375-2390.
- Jaffe E, Arber DA and Campo E. (2017), Hematopathology, 2nd Edition. Elsevier, Philadelphia. p 447-460.