Information Links
Related Conferences
Previous Issues Volume 7, Issue 4 - 2023
Investigating the Potential of Nilotinib and its Derivatives in Targeting Tau Protein Hyperphosphorylation for Alzheimer's Disease Treatment
Aria Hajihassan1, Atena Zandi1, Ghazal Esmaeili1, Mostafa Norizadeh2,3,*
1Department of Microbiology, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran
2Department of Biotechnology, Faulty of Advanced Sciences and Technology, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran
3Applied Biotechnology Research Center, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran
*Corresponding author: Mostafa Norizadeh, Department of Biotechnology, Faulty of Advanced Sciences and Technology, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran, Tel: +905372891027, E-mail: [email protected]
Received Date: November 27, 2023
Published Date: December 11, 2023
Citation: Hajihassan A, et al. (2023). Investigating the Potential of Nilotinib and its Derivatives in Targeting Tau Protein Hyperphosphorylation for Alzheimer's Disease Treatment. Mathews J Pharma Sci. 7(4):25.
Copyrights: Hajihassan A, et al. © (2023).
ABSTRACT
Alzheimer's disease, a progressive neurological disorder, significantly impacts cognitive function, memory, and behavior. Its pathogenesis is complex, involving genetic, environmental, and lifestyle factors. The tau protein, essential for axonal structure stability, is implicated in Alzheimer's disease through its role in neurofilament tangle formation. This study was aimed to investigate Nilotinib, as a tau hyperphosphorylation inhibitor, which has been demonstrated promise in treating neurodegenerative diseases, including Alzheimer’s disease, Parkinson's disease, and dementia with Lewy bodies. Tau protein structure was enhanced and molecular dynamics simulations were conducted—subsequent steps involved energy minimization and converting Nilotinib into five distinct ligands, assessing their 3D structures. To assess these ligands' interaction with the tau protein, the toxicity prediction and used site-specific molecular docking were checked. The optimal 3D structures of ligands were identified and validated for similarity to the primary macromolecule, ensuring accuracy. Docking results indicated vital binding energy and affinity of the designed ligands to the tau protein. Additionally, toxicity evaluations revealed minimal adverse effects, suggesting safety for further exploration. Preliminary findings suggest that Nilotinib and its derivatives could target tau protein hyper phosphorylation sites effectively. These results highlight the potential of Nilotinib as a therapeutic agent for treating Alzheimer's disease. However, further research is necessary to confirm these findings and evaluate the clinical applicability of Nilotinib in Alzheimer's disease treatment.
Keywords: Alzheimer, Tau, Nilotinib, Docking, Hyperphosphorylation.
INTRODUCTION
Alzheimer's disease (AD) is a neurodegenerative disorder that progresses gradually over time, resulting in a decreased ability to learn and remember. It is the primary cause of dementia in late adulthood and is linked to a high social burden as well as higher rates of death in the elderly [1,2]. AD is the most usual form of dementia, which Dr. Alois Alzheimer, a German neuropathologist, first identified in a 51-year-old woman. The demand for better medication is important because the disease’s incidence has been rising globally in recent years. Current medications have a range of adverse effects, and research is being done on natural sources of pharmaceuticals [3]. AD was the cause of around 10% of the deaths in the US in 2019. By 2050, there will be twice as many older people with AD in the US_12.7 million_as there were in 2019 (6.2 million cases estimated) [4]. Although much remains unknown about the risk factors for AD, age is the most significant element. According to the Alzheimer's Association, approximately 81% of those afflicted with AD are aged 75 years or above [5]. Genetic inheritance based on family history can also influence one's likelihood of developing the disease. Other risk factors include exposure to aluminum, traumatic brain injury (TBI), infections, and vascular disease [6,7].
Tau is considered as one of the significant microtubule-associated proteins (MAP) that binds and stabilizes microtubules with preference. Tau plays a crucial role in the axonal structure’s stability, dynamics, and maintenance. Furthermore, Tau protein’s structure and function have been linked to a number of neurological disorders, most notably Alzheimer’s disease [8,9]. Neurofibrillary tangles (NFT) and senile plaques in AD patients’ brains are the two distinct features of the disease. Amyloid beta peptide aggregations, which are produced from amyloid precursor protein (APP) are the cause of senile plaques. Tau proteins, which are mostly responsible for stabilizing microtubules in the brain, make up neurofibrillary tangles. Six different isoforms of tau can be produced in the adult human central nervous system by alternative splicing of the 16-exon MAPT gene, which is found on chromosome 17q21.3 [10,11].
Due to the presence or lack of particular exons, six isoforms exhibit variations in their core structure. Additionally, extra tau isoforms with an additional exon, exon 4a, exist in the peripheral nervous system [12]. The exact processes by which tau causes neurodegeneration in tauopathies, the role of phosphorylation and other PTMs in tau aggregation, the pathology's toxicity, and the characteristics of the toxic species are still poorly known [13]. A substantial amount of research suggests that tau hyperphosphorylation results from disturbance of cellular signaling, mostly due to an imbalance in the activity of several protein kinases and phosphatases. There are some additional functions for tau, for instance by stabilizing beta-catenin tau phosphorylation allows neurons to avoid an abrupt apoptotic death [9].
The discoveries illustrate that hyperactivation of phosphatases in normal tau can cause pair helical filaments (PHFS) and neurofibrillary tangles (NFTs) in AD patients It has been clearly demonstrated that six residual tau fragment regions, specifically PHF6 (VQIVYK) and PHF6 (VQIINK), can form PHF tau aggregates in AD patients. Several other residue positions such as Ser285, Ser289, Ser293, Ser305, and Tyr310, located near the C-terminus of the PHF6 sequence, play key roles in tau phosphorylation other tau sites participate in AD by disturbing kinase metabolism by activating phosphorylase kinase (PK) [14,15].
Tau kinases include Proline-directed kinases, glycogen synthase kinase-3 (GSK-3), cyclin-dependent kinase 5 (cdk5), and 5' adenosine monophosphate-activated protein kinase (AMPK), Non- proline-directed kinases, such as casein kinase 1 (CK1). Since tau protein dysfunction has been found to have a stronger correlation with dementia than amyloid, investigations are currently being done to find out whether targeting tau protein dysfunction can restore cognitive function in cases of Alzheimer's disease. It appears that Nilotinib penetrates the blood-brain barrier and causes autophagy in neurons to eliminate amyloid and tau protein [16].
Oral Abl tyrosine kinase inhibitor, Nilotinib, has shown promise as a disease-modifying therapy for nucleinopathies, including Alzheimer’s disease, Parkinson's disease, and dementia with Lewy bodies [17]. Additionally, research has revealed its ability to promote the autophagic elimination of a multitude of proteins that are associated with the progression of neurodegenerative diseases, including tau protein [18,19]. The medication can cause myelosuppression and has a black-box warning because of the possibility of sudden cardiac arrhythmia death. In cultured hepatic and α-synuclein overexpressing Neuro2A cells, Nilotinib has been observed to increase toxicity regardless of its ability to induce autophagy and promote α-synuclein clearance by inhibiting Abl kinase [20]. The current work examines the interactions between five different derivatives of nilotinib and the serine residues of the tau protein. The study determined the most effective tau protein binders that have inhibitory affects against tau protein aggregation through thorough in silico analysis. Mechanistic insights into the binding processes of these drugs were obtained by the simulations, which were based on protein-ligand docking techniques and optimizations using molecular mechanics. Overall, the results of this study have significant implications for the development of novel inhibitors for preventing tau protein aggregation in neurodegenerative disorders.
MATERIAL AND METHOD
Protein preparations
The UniProt database was utilized to generate a 3D structure of the human tau protein using chain A tau monomer (PDB ID: 2MZ7) within the 267-312 nucleotide range. Moreover, the structure of Tau was evaluated by PDBsum and the residues, helices and beta turns were evaluated [21] (Figure 1), Using 3Drefine [22], the desired protein was refined. Further docking preparations were carried out using Chimera v1.17.1, which involved removing excess solvents and non-complexed ions, adding polar hydrogens and atom charges, and evaluating the docking simulations. Energy minimization was performed by Chiron [23], and the final data was saved in Pdbqt format.
Ligand preparations
To begin, the molecular structure of Nilotinib (PubChemCID_644241) was obtained from the PubChem database in sdf format. The primary ligand was then derived using Chem Draw v22.2.0. Five derivatives were designed using Chem3D v22.2.0 and their three-dimensional structures were generated as potential inhibitors of tau protein aggregation (Table 1). The ligands were optimized with MM2 Job command, minimizing the molecular energy. Afterwards, molecular dynamics simulations were performed to analyze the most promising structures at a temperature of 300 Kelvin. To assess the quality of the resulting structures, web-based interaction analysis and structural evaluations were employed.
Determination of docking coordinates
According to previous studies, optimal residues (SER285 - SER289 - SER293) which play key roles in tau phosphorylation were selected. Subsequently, the grid box with covering coordinates (x = 17.85, y = -4.988, z = -25.871) were defined by Autodock Tool 4.2 to determine the most appropriate range to start specific molecular docking process.
Molecular Docking
The goal of ligand—protein docking, which is an analytical descriptive procedure, is to predict the predominant binding model(s) of a ligand with a protein of known 3D structure [24]. In order to start docking procedure, high-dimensional spaces were analyzed and scoring function was used, which correctly ranked docking candidates, which was performed by Autodock Vina [25]. The desired ligands were then defined within the software. The docking results were subsequently categorized based on the top outcome.
Toxicity prediction
First, the ligands were checked by SwissADME [26] and the amounts of lipophility, molecular size, polarity, insolubility, instauration and flexibility were checked. ProTox-II server was used to detect the level of compound toxicity and determine ADMET (absorption, distribution, metabolism, excretion, and toxicity) variables for Nilotinib as well as it’s designed derivatives. The Final Data was utilized for prediction of multiple toxicological endpoints related with a chemical structure. In this study, toxicity class predicted LD50 and possible clinical complications were investigated [27-29].
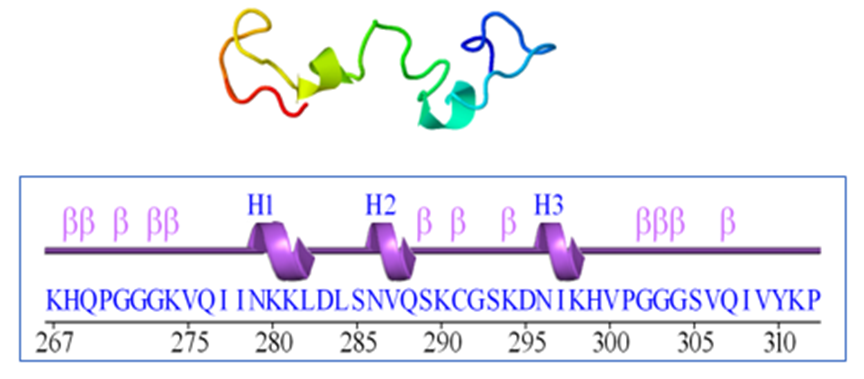
Figure 1. The 3D structure of the 2mz7 protein is shown above and the two-dimensional structure including residue 46 below includes 3 helices and 12 beta turns (B turns) and no gamma turns.
Table 1. Ligands derived from Nilotinib by Chemdraw along with chemical formula, Number of bonds, molecular weight and m/z (mass-to-charge ratio)
.PNG)
RESULTS
Based on the comparison of all five proposed protein structures with the 3Drefine server, the results were obtained: Through analysis Scores, it was determined that structure number three had the most optimal conformation. This structure exhibited a lower RW plus index and MolProbity index, indicating a more accurate representation of the molecule's geometry (Table 2). After selecting the protein (which was the 3th model), the clashes and possible collisions between the residues of the final protein with the help of the Chiron server in order to reach the lowest numbers of clashes were checked (Table 3). Analyzed by Procheck [30], the z score index is equal to-0.57, indicating the degree of proximity of the selected protein in the range of index proteins in the same number of residues (Figure 2). The viability of the selected model by visualizing the Ramachandran plot and comparing the third indexed residue to all 118 indexed residues were proved (Figure 3).
As a result of docking according to table x for ligands (a_f) binding affinity was obtained from -5.2 to -8.5, which is the best result for ligand c. The results with rmsd equal to zero for ligand a equal to -5.7, ligand b equal to -7.3, for ligand c equal to -8.5, for ligand d equal to -6.7, for ligand e equal to -7.4 and for ligand f equal to - 7.4 has been obtained (Table 4). In the next step, the data was displayed graphically (Figure 4). The Discovery Studio software was used to predict the molecular interactions between a ligand and a protein. The results showed that Nilotinib formed halogen bonds with serine 289, serine 288, and cysteine 291. Structure B formed a hydrogen bond with leucine 282 and aspartic acid 283. Structure C established hydrogen bonds with leucine 282, aspartic acid 283, and serine 293 in structure D, halogen bonds were formed with glutamine 288, glutamine 269, cysteine 291, and histidine 268, as well as hydrogen bonds with aspartic acid 283 and serine 285. It also formed unfavorable donor-donor bond with Glutamic acid 288 and carbon hydrogen bond with Serin 293. Structure F had hydrogen bonds with leucine 284 and halogen bonds with glycine 292 and glutamine 288 (Figure 5). Swiss results showed that Nilotinib and its derivatives had a degree of unsaturation higher than the optimal level. Flexibility, lipophilicity and polarity of ligands have a standard limit. Both C and D ligands showed better chemical properties (Figure 6). According to the prediction obtained by the ProtoTox-II server, the amount of LD Nilotinib of structure B and structure was 800. The highest LD and, as a result, the lowest lethality risk related to D structure was reported with the number 2500. The toxicity class for all ligands except D showed the number 4, while the structure of D was placed in class 5 (Figure 7). The classification of ligands showed that all the drugs were hepatotoxicin, and this problem was less severe in B, D and F drugs. Unlike D and B, nilotinib, C, E and F were carcinogenic. The structures of CF mutagenicity and H receptors were active. Ultimately, all drugs showed immunotoxicity (Table 5).
Table 2. The refinement results obtained from the 3Drefine server - Model number three got the best score
|
Model |
3Drefine Score |
RWPlus |
|
3 |
3840.83 |
-3931.022438 |
|
4 |
3814.58 |
-3924.925192 |
|
5 |
3789.66 |
-3916.572491 |
|
2 |
3894.85 |
-3912.910533 |
|
1 |
4057.01 |
-3911.202419 |
|
Atom1 |
Residue1 |
Atom2 |
Residue2 |
Accepted Distance (Å) |
Actual Distance (Å) |
VDW Repulsion (kcal/mol) |
|
A1 |
R1 |
A2 |
R2 |
Acc.D |
Act.D |
VDWR |
|
CA |
1 |
CD |
14 |
4.425 |
3.561 |
0.498 |
|
C |
2 |
CD |
4 |
4.16 |
3.422 |
0.326 |
|
CD |
3 |
CA |
19 |
4.465 |
3.492 |
0.403 |
|
OE1 |
3 |
CA |
19 |
3.965 |
3.135 |
0.608 |
|
O |
22 |
N |
24 |
3.2 |
2.629 |
0.782 |
|
O |
29 |
CA |
32 |
3.965 |
3.26 |
0.35 |
|
O |
30 |
N |
32 |
3.2 |
2.451 |
1.8 |
|
CB |
33 |
CG |
46 |
4.119 |
3.429 |
0.346 |
|
CB |
33 |
CD |
46 |
4.119 |
3.331 |
0.599 |
|
O |
34 |
CG1 |
42 |
3.66 |
3.19 |
0.413 |
|
O |
42 |
N |
44 |
3.2 |
2.611 |
0.885 |
|
CD1 |
42 |
CE1 |
44 |
3.998 |
3.259 |
0.677 |
.png)
(1).png)
Figure 2. The ProSA z score of -0.57 was found within the characteristics range of native protein conformation, indicating that the protein structure had extremely few errors. The blue color is the least absorbent energy and the red color represents the most repulsive energy.
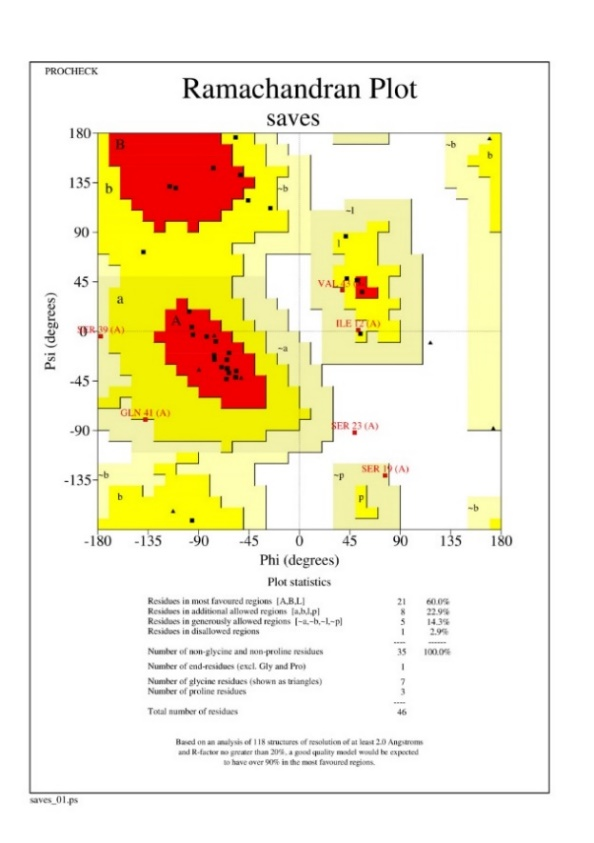
Figure 3. Ramachandran is plotted for all 46 residues. Residues in the red areas (A, B, L) are the most consistent and the residues in the yellow areas are acceptable. Finally, 96.3% of the residues without glycine and proline were acceptable.
Table 4. The results obtained from Autodock Vina- Nilotinib and its derivatives reacted with protein and the results were reported by binding affinity and Rmsd/lower bound–upper bound
|
Rank |
Binding Affinity |
rmsd/ub |
rmsd/lb |
|
1 |
-5.7 |
0 |
0 |
|
2 |
-5.7 |
5.845 |
1.515 |
|
3 |
-5.6 |
3.167 |
0.695 |
|
4 |
-5.5 |
4.983 |
1.359 |
|
5 |
-5.4 |
3.721 |
1.462 |
|
6 |
-5.3 |
3.466 |
1.399 |
|
7 |
-5.3 |
4.449 |
2.334 |
|
8 |
-5.2 |
12.406 |
10.057 |
|
9 |
-5.2 |
4.981 |
2.602 |
|
Rank |
Binding Affinity |
rmsd/ub |
rmsd/lb |
|
1 |
-7.3 |
0 |
0 |
|
2 |
-7.2 |
1.684 |
1.466 |
|
3 |
-7.2 |
2.68 |
2.283 |
|
4 |
-7.1 |
10.122 |
4.684 |
|
5 |
-7 |
13.052 |
8.637 |
|
6 |
-6.9 |
10.327 |
4.838 |
|
7 |
-6.8 |
10.337 |
4.86 |
|
8 |
-6.7 |
10.383 |
5.297 |
|
9 |
-6.6 |
11.229 |
6.287 |
|
Rank |
Binding Affinity |
rmsd/ub |
rmsd/lb |
|
1 |
-8.5 |
0 |
0 |
|
2 |
-8.4 |
10.586 |
4.949 |
|
3 |
-8.1 |
9.808 |
4.566 |
|
4 |
-8.1 |
10.946 |
4.525 |
|
5 |
-8.1 |
4.045 |
1.728 |
|
6 |
-8.1 |
6.522 |
3.596 |
|
7 |
-7.9 |
10.865 |
5.1 |
|
8 |
-7.9 |
12.393 |
8.266 |
|
9 |
-7.8 |
4.504 |
2.792 |
|
Rank |
Binding Affinity |
rmsd/ub |
rmsd/lb |
|
1 |
-6.8 |
0 |
0 |
|
2 |
-6.5 |
2.459 |
1.97 |
|
3 |
-6.3 |
3.391 |
2.011 |
|
4 |
-6.3 |
3.045 |
2.308 |
|
5 |
-6.2 |
3.727 |
2.508 |
|
6 |
-6.1 |
9.539 |
2.573 |
|
7 |
-6 |
11.457 |
7.976 |
|
8 |
-6 |
2.799 |
1.904 |
|
9 |
-5.9 |
3.163 |
2.168 |
|
Rank |
Binding Affinity |
rmsd/ub |
rmsd/lb |
|
1 |
-7.4 |
0 |
0 |
|
2 |
-7.4 |
9.718 |
4.267 |
|
3 |
-7.4 |
9.981 |
4.118 |
|
4 |
-7.4 |
9.458 |
4.06 |
|
5 |
-7.3 |
12.498 |
8.333 |
|
6 |
-7.3 |
7.815 |
3.124 |
|
7 |
-7.2 |
9.16 |
4.094 |
|
8 |
-7.2 |
3.422 |
2.238 |
|
9 |
-7.2 |
12.507 |
6.293 |
|
Rank |
Binding Affinity |
rmsd/ub |
rmsd/lb |
|
1 |
-7.4 |
0 |
0 |
|
2 |
-7.3 |
10.785 |
3.877 |
|
3 |
-7.1 |
13.178 |
8.141 |
|
4 |
-7.1 |
10.971 |
4.247 |
|
5 |
-7 |
13.067 |
8.539 |
|
6 |
-6.9 |
11.062 |
3.561 |
|
7 |
-6.9 |
15.083 |
8.196 |
|
8 |
-6.9 |
10.322 |
4.989 |
|
9 |
-6.7 |
11.267 |
3.751 |
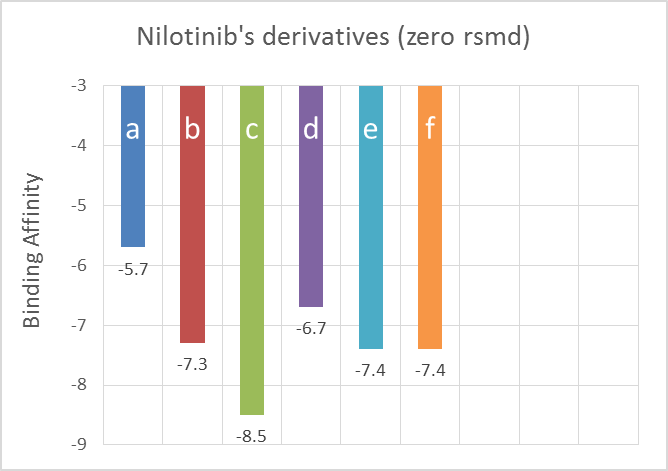
Figure 4. Data obtained from AutoDock Vina- Ranking shows binding affinity for Nilotinib and its derivatives with zero rmsd.
.png)
.png)
Nilotinib (a)
.png)
.png)
Nilotinib (b)
.png)
.png)
(c)
.png)
.png)
(d)
.png)
.png)
(e)
.png)
.png)
(f)
Figure 5. The Docking simulation conducted by Discovery Studio shows 3D protein-ligand interactions (left figures) / 2D protein-ligand interaction maps (right figures)
(1).png)
Nilotinib (a)
.png)
(b)
(1).png)
(c)
(1).png)
(d)
(1).png)
(e)
(1).png)
(f)
Figure 6. Chemical properties: the ideal range of each compound for each property is shown by the pink area. The diagram displays the following properties: flexibility (FLEX), unsaturation (INSATU), polarity (POLAR), insolubility (INSOLU), and lipophilicity (LIPO).
.png)
Nilotinib (a)
.png)
(b)
.png)
(c)
.png)
(d)
.png)
(e)
.png)
(f)
Figure 7. Ligands’ toxicity rankings, using ProtoTox-II server, compared to known drugs
(LD50= median lethal dose).
Table 5. Toxicity predictions performed by ProtoTox-II, indicating toxic classification and target probabilities
|
Classification |
Target |
Shorthand |
Prediction |
Probability |
|
Organ toxicity |
Hepatotoxicity |
dili |
Active |
0.82 |
|
Toxicity end points |
Carcinogenicity |
carcino |
Active |
0.53 |
|
Toxicity end points |
Immunotoxicity |
immuno |
Active |
0.98 |
|
Toxicity end points |
Mutagenicity |
mutagen |
Inactive |
0.59 |
|
Toxicity end points |
Cytotoxicity |
cyto |
Inactive |
0.72 |
|
Tox21-Nuclear receptor signaling pathways |
Aryl hydrocarbon Receptor (AhR) |
nr_ahr |
Inactive |
0.57 |
|
Classification |
Target |
Shorthand |
Prediction |
Probability |
|
Organ toxicity |
Hepatotoxicity |
dili |
Active |
0.64 |
|
Toxicity end points |
Carcinogenicity |
carcino |
Inactive |
0.58 |
|
Toxicity end points |
Immunotoxicity |
immuno |
Active |
0.99 |
|
Toxicity end points |
Mutagenicity |
mutagen |
Inactive |
0.66 |
|
Toxicity end points |
Cytotoxicity |
cyto |
Inactive |
0.69 |
|
Tox21-Nuclear receptor signaling pathways |
Aryl hydrocarbon Receptor (AhR) |
nr_ahr |
Inactive |
0.55 |
|
Classification |
Target |
Shorthand |
Prediction |
Probability |
|
Organ toxicity |
Hepatotoxicity |
dili |
Active |
0.78 |
|
Toxicity end points |
Carcinogenicity |
carcino |
Active |
0.58 |
|
Toxicity end points |
Immunotoxicity |
immuno |
Active |
0.99 |
|
Toxicity end points |
Mutagenicity |
mutagen |
Active |
0.63 |
|
Toxicity end points |
Cytotoxicity |
cyto |
Inactive |
0.72 |
|
Tox21-Nuclear receptor signaling pathways |
Aryl hydrocarbon Receptor (AhR) |
nr_ahr |
Active |
0.51 |
|
Classification |
Target |
Shorthand |
Prediction |
Probability |
|
Organ toxicity |
Hepatotoxicity |
dili |
Active |
0.64 |
|
Toxicity end points |
Carcinogenicity |
carcino |
Inactive |
0.52 |
|
Toxicity end points |
Immunotoxicity |
immuno |
Active |
0.99 |
|
Toxicity end points |
Mutagenicity |
mutagen |
Inactive |
0.69 |
|
Toxicity end points |
Cytotoxicity |
cyto |
Inactive |
0.68 |
|
Tox21-Nuclear receptor signaling pathways |
Aryl hydrocarbon Receptor (AhR) |
nr_ahr |
Inactive |
0.72 |
|
Classification |
Target |
Shorthand |
Prediction |
Probability |
|
Organ toxicity |
Hepatotoxicity |
dili |
Active |
0.82 |
|
Toxicity end points |
Carcinogenicity |
carcino |
Active |
0.53 |
|
Toxicity end points |
Immunotoxicity |
immuno |
Active |
0.98 |
|
Toxicity end points |
Mutagenicity |
mutagen |
Inactive |
0.59 |
|
Toxicity end points |
Cytotoxicity |
cyto |
Inactive |
0.72 |
|
Tox21-Nuclear receptor signaling pathways |
Aryl hydrocarbon Receptor (AhR) |
nr_ahr |
Inactive |
0.57 |
|
Classification |
Target |
Shorthand |
Prediction |
Probability |
|
Organ toxicity |
Hepatotoxicity |
dili |
Active |
0.61 |
|
Toxicity end points |
Carcinogenicity |
carcino |
Active |
0.62 |
|
Toxicity end points |
Immunotoxicity |
immuno |
Active |
0.99 |
|
Toxicity end points |
Mutagenicity |
mutagen |
Active |
0.75 |
|
Toxicity end points |
Cytotoxicity |
cyto |
Inactive |
0.61 |
|
Tox21-Nuclear receptor signaling pathways |
Aryl hydrocarbon Receptor (AhR) |
nr_ahr |
Active |
0.64 |
DISCUSSION
This study focused on investigating the tau protein and developing five engineered ligands to interact with specific serine residues. The objective was to find compound candidates that can inhibit tau protein aggregation, which is associated with neurodegenerative diseases like Alzheimer's. Based on the examination of existing scientific literature, it has been observed that a general augmentation in the process of tau phosphorylation leads to a decrease in its binding strength with microtubules, thereby resulting in the destabilization of the neuronal cytoskeleton [31, 32]. In recent scientific investigations, a considerable number of studies have been conducted to explore the inhibitory effects of newly developed compounds. Notably, research examining Curcumin and its derivatives has demonstrated a notable affinity for binding with the tau microtubule association protein. However, it is important to note that studies involving high doses of curcumin have reported mild adverse reactions, including symptoms such as nausea, diarrhea, headache, skin rash, and the presence of yellow-colored stool [33,34]. In another study focused on the binding of donepezil to the Tau protein, researchers employed both surface plasmon resonance (SPR) and molecular modeling methodologies. The findings revealed that the interaction between these compounds is influenced by both hydrogen bonding and van der Waals forces, as indicated by the presence of negative values for enthalpy and entropy. It is important to note that common side effects of donepezil administration include diarrhea, headaches, and nausea [35,36]. The findings revealed that one of the ligands, called "c," had the strongest binding affinity to serine 293 of the tau protein. This serine residue is known to be involved in hyper phosphorylation, supporting the significance of the findings. Ligand "c" showed a pronounced ability to engage with the tau protein and hinder aggregation in that specific domain.
However, an important observation regarding ligand "c" was its considerable carcinogenic, mutagenic, and hepatotoxic properties. It also had a high predicted LD50 value of 1000 mg/kg, indicating safety concerns. On the other hand, ligand "b" ranked second in the docking analysis and exhibited lower toxicity compared to the other compounds. It still had immunotoxic and hepatotoxic effects, with a predicted LD50 value of 800 mg/kg. Ligands "e" and "f" ranked third based on their docking scores, with ligand "f" interacting with serine 293 and having a toxicity profile similar to ligand "c." Ligand "d" occupied the subsequent position and showed reduced toxicity and lethality compared to nilotinib and the other ligands. Notably, it strongly interacted with serine 285 and demonstrated higher flexibility compared to the other ligands. Ligand "D" fell within the fifth category of drug toxicity, indicating an acceptable safety profile, with a predicted LD50 value of 2500 mg/kg. Lastly, ligand "a" (Nilotinib) ranked the lowest in the docking analysis. Although its binding affinity was not as high as the others, it had a tolerable toxicity profile, with an LD50 value of 800 mg/kg. It formed a halogen bond with serine 285 and serine 289, making it a potentially less toxic alternative. In conclusion, ligand "c" showed remarkable promise in terms of binding affinity but had significant toxicity issues. Conversely, ligand "d" emerged as a compelling candidate with reduced toxicity and better flexibility. Ligand "a" (Nilotinib), despite ranking lowest, exhibited prospective attributes, especially regarding its safety profile. This analysis raises questions about the complex puzzle that lies ahead.
CONCLUSION
Our results exhibited the top five ligand molecules, showing binding affinity with the tau receptor site through molecular docking energies and protein-ligand molecular interactions such as hydrogen bonding and halogen bond interactions. By examining the level of toxicity, it can be concluded that the structure of (d) with the lowest risk and acceptable binding to serine is a suitable ligand to cover this area and prevent the activity of kinase enzymes and the subsequent absence of hyperphosphorylation. As a result, this drug can be studied in vitro, in vivo and in clinical trials to help treating Alzheimer's disease.
REFERENCES
- Cass SP. (2017). Alzheimer's Disease and Exercise: A Literature Review. Curr Sports Med Rep. 16(1):19-22.
- Thal DR, Tomé SO. (2022). The central role of tau in Alzheimer's disease: From neurofibrillary tangle maturation to the induction of cell death. Brain Res Bull. 190:204-217.
- Abraham JT, Maharifa HNS, Hemalatha S. (2022). In Silico Molecular Docking Approach Against Enzymes Causing Alzheimer's Disease Using Borassus flabellifer Linn. Appl Biochem Biotechnol. 194(4):1804-1813.
- Ho JY, Franco Y. (2022). The rising burden of Alzheimer's disease mortality in rural America. SSM Popul Health. 17:101052.
- Pradeepkiran JA, Reddy PH. (2019). Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer's Disease. Cells. 8(3):260.
- A Armstrong R. (2019). Risk factors for Alzheimer's disease. Folia Neuropathol. 57(2):87-105.
- Mold M, Linhart C, Gómez-Ramírez J, Villegas-Lanau A, Exley C. (2020). Aluminum and Amyloid-β in Familial Alzheimer's Disease. J Alzheimers Dis. 73(4):1627-1635.
- Dixit H, Kumar CS, Chaudhary R, Thaker D, Gadewal N, Dasgupta D. (2021). Role of Phosphorylation and Hyperphosphorylation of Tau in Its Interaction with βα Dimeric Tubulin Studied from a Bioinformatics Perspective. Avicenna J Med Biotechnol. 13(1):24-34.
- Medeiros R, Baglietto-Vargas D, LaFerla FM. (2011). The role of tau in Alzheimer's disease and related disorders. CNS Neurosci Ther. 17(5):514-524.
- Noble W, Hanger DP, Miller CC, Lovestone S. (2013). The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol. 4:83.
- Sadikoglou E, Domingo-Fernández D, Savytska N, et al. (2022). Rescue of the increased susceptibility to Mild Chronic Oxidative Stress of iNeurons carrying the MAPT Chromosome 17q21.3 H1/H1 risk allele by FDA-approved compounds. bioRxiv. DOI: 10.1101/2022.11.07.515284.
- Avila J, Jiménez JS, Sayas CL, Bolós M, Zabala JC, Rivas G, et al. (2016). Tau Structures. Front Aging Neurosci. 8:262.
- Haj-Yahya M, Gopinath P, Rajasekhar K, Mirbaha H, Diamond MI, Lashuel HA. (2019). Site-specific hyperphosphorylation of tau inhibits its fibrillization in vitro, blocks its seeding capacity in cells, and disrupts its microtubule binding; Implications for the native state stabilization of tau. bioRxiv. pp. 772046.
- Inoue M, Konno T, Tainaka K, Nakata E, Yoshida HO, Morii T. (2012). Positional effects of phosphorylation on the stability and morphology of tau-related amyloid fibrils. Biochemistry. 51(7):1396-1406.
- Man VH, He X, Han F, Cai L, Wang L, Niu T, Zhai J, Ji B, Gao J, Wang J. (2023). Phosphorylation at Ser289 Enhances the Oligomerization of Tau Repeat R2. J Chem Inf Model. 63(4):1351-1361.
- Pluta R, Ułamek-Kozioł M. (2020). Tau protein-targeted therapies in Alzheimer’s disease: current state and future perspectives. Australia: Exon Publications. p. 69-82.
- Turner RS, Hebron ML, Lawler A, Mundel EE, Yusuf N, Starr JN, et al. (2020). Nilotinib Effects on Safety, Tolerability, and Biomarkers in Alzheimer's Disease. Ann Neurol. 88(1):183-194.
- Lonskaya I, Hebron ML, Desforges NM, Schachter JB, Moussa CE. (2014). Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J Mol Med (Berl). 92(4):373-386.
- Hamano T, Enomoto S, Shirafuji N, Ikawa M, Yamamura O, Yen SH, et al. (2021). Autophagy and Tau Protein. Int J Mol Sci. 22(14):7475.
- Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C, et al. (2010). Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 362(24):2251-2259.
- Laskowski RA, Jabłońska J, Pravda L, Vařeková RS, Thornton JM. (2018). PDBsum: Structural summaries of PDB entries. Protein Sci. 27(1):129-134.
- Bhattacharya D, Nowotny J, Cao R, Cheng J. (2016). 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Res. 44(W1):W406-W409.
- Ramachandran S, Kota P, Ding F, Dokholyan NV. (2011). Automated minimization of steric clashes in protein structures. Proteins. 79(1):261-270.
- Leach AR, Shoichet BK, Peishoff CE. (2006). Prediction of protein-ligand interactions. Docking and scoring: successes and gaps. J Med Chem. 49(20):5851-5855.
- Trott O, Olson AJ. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 31(2):455-461.
- Daina A, Michielin O, Zoete V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 7:42717.
- Banerjee P, Dehnbostel FO, Preissner R. (2018). Prediction Is a Balancing Act: Importance of Sampling Methods to Balance Sensitivity and Specificity of Predictive Models Based on Imbalanced Chemical Data Sets. Front Chem. 6:362.
- Banerjee P, Eckert AO, Schrey AK, Preissner R. (2018). ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 46(W1):W257-W263.
- Drwal MN, Banerjee P, Dunkel M, Wettig MR, Preissner R. (2014). ProTox: a web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 42(Web Server issue):W53-W58.
- Laskowski RA, MacArthur MW, Thornton JM. (2012). PROCHECK: validation of protein-structure coordinates. International Tables for Crystallography. F(ch. 21.4):684-687.
- Hutton M. (2001). Missense and splice site mutations in tau associated with FTDP-17: multiple pathogenic mechanisms. Neurology. 56(11 Suppl 4):S21-S25.
- Eidenmüller J, Fath T, Hellwig A, Reed J, Sontag E, Brandt R. (2000). Structural and functional implications of tau hyperphosphorylation: information from phosphorylation-mimicking mutated tau proteins. Biochemistry. 39(43):13166-13175.
- Burgos-Morón E, Calderón-Montaño JM, Salvador J, Robles A, López-Lázaro M. (2010). The dark side of curcumin. Int J Cancer. 126(7):1771-1775.
- Cianfruglia L, Minnelli C, Laudadio E, Scirè A, Armeni T. (2019). Side Effects of Curcumin: Epigenetic and Antiproliferative Implications for Normal Dermal Fibroblast and Breast Cancer Cells. Antioxidants (Basel). 8(9):382.
- Jackson S, Ham RJ, Wilkinson D. (2004). The safety and tolerability of donepezil in patients with Alzheimer's disease. Br J Clin Pharmacol. 58 Suppl 1(Suppl 1):1-8.
- Seltzer B. (2005). Donepezil: a review. Expert Opin Drug Metab Toxicol. 1(3):527-536.