Information Links
Related Conferences
Previous Issues Volume 7, Issue 1 - 2023
A Review on Formulation and Evaluation Approaches for Fast Release Tablet
Prashant Upadhyay*, Kalpana, Priya Chaudhary, Sukirti Upadhyay
School of Pharmaceutical Sciences, IFTM University, Moradabad, Uttar Pradesh, India *Corresponding author: Prashant Upadhyay, School of Pharmaceutical Sciences, IFTM University, Moradabad, Uttar Pradesh, India; Tel: 9719915567; Email: [email protected]
Received Date: 09 Jan 2023
Published Date: 25 Jan 2023
Citation: Upadhyay P, et al. (2023). A Review on Formulation and Evaluation Approaches for Fast Release Tablet. Mathews J Pharma Sci. 7(1):15.
Copyrights: Upadhyay P, et al. © (2023).
ABSTRACT
Among all forms of volume, the tablet is the most popular volume method available today due to its convenience of self-control, coherence and easy production; sometimes immediate relief the onset of action is necessary than conventional treatment in most cases. To overcome these barriers, a form of immediate release dose has emerged like other oral dose forms. Forms of rapid drug release dispersal immediately after treatment with an improved degree of elimination. The basic method used in pill development is the use of superdisintegrants such as Kyron T-314 based on polymer bonded Polycarboxylic acid per USP/NF and has a K+ionic form. The Kyron T-316 is a relatively new fast disintegration system specifically for acid-containing drugs that promote dispersion and dissolution. Sodium starch glycolate (Primogel, Explotab), carboxymethyl cellulose (Croscarmellose) etc. The drug categories selected for immediate release are painkillers and anti-inflammatory drugs such as Ibuprofen, Diclofenac sodium. Anti-coagulants such as Dicoumarol, Dipyridamol. Anti-Depressants like Amoxapiine. Antidiabetic as Glipizide. Antibacterial like Linezolid, antihypertensive drugs like Amlodipine, Minoxidil, Nifedipine are best for immediate release.
Keywords: Fast release, Polymers, Super disintegrants, Kyron T-314 And 316, Wet Granulation, Release Kinetics
INTRODUCTION
The oral method is the most commonly used method of drug administration. Although a different route of administration is used for drug delivery, due to the flexibility in the formulation of the dosage form and oral method of patient compliance is preferred [1]. The popularity of oral transmission is said to be easy to manage, patient acceptance, accurate dosage, low cost production method and improved product shelf life [2].
There are several ways to make a drug delivery system where pills are used as drug carriers. Among them, a solid structure does not require sterile conditions and therefore, is less expensive to pick [3].
The tablet is the most widely used volume method due to its convenience of self-control, coherence and ease of production [4]. Tablets are solid dosage forms containing therapeutic substances with or without cleaners [5]. According to Indian Pharmacopoeia herbal pills are solid, flat or biconvex containers, a unit-standard form, prepared by pressing a drug or drug mixture, with or without diluent [6].
The fast-release pills are the ones that disperse quickly and dissolve to release drugs [7]. With the construction of rapid release, superdisintegrants play an important role. Superdisintegrants are used to improve the performance of a solid volume form. This is achieved in a variety of ways, inflammation, porosity and capillary action, moisturizing temperature, particle expulsion capacity, regeneration, enzymatic reactions when pills are broken down into particles [8]. Fast-dispersing pills (FDTs) have found a growing demand over the past decade, and the field has become a fast-growing area in the pharmaceutical industry. The popularity and usefulness of construction has resulted in the development of many FDT technologies.
Fast-acting pills (FDTs) are very popular all over the world. Based on requests from patients to improve their quality of life (QOL), new types of FDT have been developed and released worldwide by many pharmaceutical companies [9]. A review study was carried out on formulation and evaluation approaches for fast release tablet.
Fast release drug delivery system benefits improved stability, bioavailability, and compliance appropriate for continuous or regulated discharge, permit heavy drug use a solid preparation's capacity to deliver the advantages of liquid medicines, Flexible and able to work with current processing and packaging machinery, improved chemical solubility at a lower cost. The drug categories that are selected for immediate release are painkillers and anti-inflammatory drugs such as Ibuprofen, Diclofenac sodium. Anti-coagulants such as Dicoumarol, Dipyridamol. Anti-Depressants like Amoxapiine. Antidiabetic as Glipizide. Anti-bacterial such as Linezolid, antihypertensive drug like Amlodipine, Minoxidil, Nifedipine are best for immediate release [10].
GENERAL EXCIPIENTS USED IN IMMEDIATE RELEASE TABLETS
Suitable features of excipient [11,12]
•They must not have an unacceptable microbiological load.
•It must match the colour; it should not change the colour shade in the construction.
•If the product is classified as food, detergents and other additives must be approved for food additives.
•They must not adversely affect the availability of organic products. They must be non-toxic and non-medical and must be approved by regulatory agencies in the countries where the product will be marketed.
• Must be commercially available at a reasonable level in the countries where the product will be produced.
•Cost effective.
•They must be physically inactive.
•They must be physically and chemically stable and combined with other medications and components of the pill.
Table 1: Excipients used in solid dosage forms.
|
EXCIPIENTS |
FUNCTION |
EXAMPLE |
|
Diluent |
Serve as bulking agents and facilitate accurate dosing. |
Sugar compounds: lactose, mannitol, dextrose, sorbitol, silicate, calcium, magnesium salt, sodium chloride, potassium chloride, cellulose derivatives |
|
Binder, compression aid, granulating agents |
Facilitate tablet compression. Ensure tablet robustness. |
Natural and synthetic polymer: starch, gelatin, and sugars as sucrose, glucose, dextrose, and lactose |
|
Disintegrants
Superdisintegrants |
Aid with tablet disintegration and dissolution by increasing the surface area of the tablets, facilitate release of drug substance. improved disintegrant efficacy resulting in decreased use levels when compared to traditional disintegrants |
Compounds which swell in the presence of water: starch, cellulose derivatives, alginates and crospovidone
Croscarmellose, cross povidone, sodium starch glycolate, polacrilin potassium |
|
Glidants |
Granulation flow enhancer, aid with tablet compression and eliminate particles agglomeration (anticaking) |
Colloidal anhydrous silicon, silica compounds, talc |
|
Lubricants |
Tablet compression aid, reduce blend cohesiveness characteristic during compression, reduce disintegration rate |
Steric acid, salts, and derivatives of steric acid, talc, hydrogenated vegetable oils and PEG |
|
Coating agent |
Prevent tablet degradation environmental conditions (temperature, light and moisture). Serve as taste masking agents, inhibit order, facilitate administration and appearance enhancer |
Natural and synthetic polymers, polymers that are insoluble in acid |
As disintegrants sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, Crospovidone, polyvinyl polypyrrolidone, methyl cellulose, microcrystalline cellulose, alkyl-substituted hydroxypropyl cellulose, alkyl-substituted hydroxypropyl cellulose, starchlin, alkyl-substituted hydroxypropyl cellulose compounds. The amount of disintegrant included in the volume form will depend on a number of factors, including dispersion structures, porosigen structures and properties of selected disintegrants. Generally, a disintegrant will cover from 1% w / w to 25% w / w of volume form [13].
Mechanism of tablet Disintegration [14]
The tablet breaks to primary particles by one or more of the mechanisms listed below:
1) By capillary action
2) By swelling
3) Because of heat of wetting
4) Due to disintegrating particle/particle repulsive force
5) Due to deformation
6) Due to release of gases
7) By enzymatic action
Super Disintegrates
A disintegrant is an excipient, which is added to a tablet or capsule blend to aid in the break-up of the compacted mass when it is put into a fluid environment.
Advantages:
- Effective in lower concentrations
- Less effect on compressibility and flowability
- More effective intragranularly
Some super disintegrants are:
1. Sodium Starch Glycolate (Explotab, Primogel) used in concentration of 2-8 % & optimum is 4%. Mechanism of Action: Rapid and extensive swelling with minimal gelling. Microcrystalline cellulose (Synonym: Avicel, celex) used in concentration of 2- 15% of tablet weight. And Water wicking.
2. Cross-linked Povidone or crospovidone (Kollidone) used in concentration of 2-5% of weight of tablet. Completely insoluble in water.
Mechanism of Action: Water wicking, swelling and possibly some deformation recovery. Rapidly disperses and swells in water, but does not gel even after prolonged exposure. Greatest rate of swelling compared to other disintegrants. Greater surface area to volume ratio than other disintegrants.
3. Low-substituted hydroxyl propyl cellulose: Which is insoluble in water. Rapidly swells in water. Grades LH-11 and LH-21 exhibit the greatest degree of swelling. Certain grades can also provide some binding properties while retaining disintegration capacity. Recommended concentration 1-5%
4. Cross linked carboxy methyl cellulose sodium (Ac-Di-sol): Croscarmellose sodium: Mechanism of Action: Wicking due to fibrous structure, swelling with minimal gelling. Effective Concentrations: 1-3% Direct Compression, 2-4% Wet Granulation [15].
5. Kyron T-314: Kyron T-314 is derived from cross-linked polymer of Polycarboxylic acids as per USP/NF & has the K+ ionic form. It is a very high purity polymer used in pharmaceutical formulations as a superfast disintegrant as well as dissolution improver in solid dosage forms like tablets, capsules, pellets etc. It is available in white free flowing powder hence it is suitable for the both wet granulation as well as direct compression system for tablet formulations.
6. Kyron T-316: Kyron T- 316 has a very high swelling tendency of hydration either in contact with water or G.I. fluids causing fast disintegration without the formation of lumps and thus acts as an effective tablet super disintegrant. It can be used during mixing and/or at lubrication stage.
Examples: fast release tablets, Herbal tablets, Mouth dissolving tablets, etc. [16].
BINDER
Binder is a material used to bind other materials together. Microcrystalline cellulose (MCC) is commonly used as a filler-binder in direct compression because of its good bonding properties. Other commonly used binders in direct compression include starches and their derivatives, such as pregelatinised and granulated starches.
SURFACTANTS
One very useful class of excipients is surfactants, preferably present from 0 to 10 % w/w. Suitable surfactants include fatty acid and alkyl sulfonates; commercial surfactants such as benzalkonium chloride, dioctyl sodium sulfosuccinate, polyoxyethylene sorbitan fatty acid esters, natural surfactants such as sodium taurocholic acid, lecithin, and other phospholipids and mono and diglycerides; and mixtures thereof. Such materials can advantageously be employed to increase the rate of dissolution by, for example, facilitating wetting, or otherwise increase the rate of drug release from the dosage form [17].
pH Modifiers
Inclusion of pH modifiers such as acids, bases, or buffers may also be beneficial in an amount of from 0 to 10 % w/w. Acidic pH modifiers (e.g., acids such as citric acid or succinic acid) retard the dissolution of the pharmaceutical composition when the dispersion polymer is anionic. Alternatively, basic pH modifiers (e.g., sodium acetate or amines) enhance the rate of dissolution of the same types of pharmaceutical composition [16].
DILUENTS
Examples of other matrix materials, fillers, or diluents include lactose, mannitol, xylitol, dextrose, sucrose, sorbitol, compressible sugar, microcrystalline cellulose (MCC), powdered cellulose, starch, pregelatinized starch, dextrates, dextran, dextrin, dextrose, maltodextrin, calcium carbonate, dibasic calcium phosphate, tribasic calcium phosphate, calcium sulfate, magnesium carbonate, magnesium oxide, poloxamers, polyethylene oxide, hydroxypropyl methyl cellulose (HPMC) and mixtures thereof [17].
LUBRICANTS
Examples of lubricants include calcium stearate, glyceryl monostearate, glyceryl palmitostearate, hydrogenated vegetable oil, light mineral oil, magnesium stearate, mineral oil, polyethylene glycol, sodium benzoate, sodium lauryl sulfate, sodium stearyl fumarate, stearic acid, talc and zinc stearate [18].
GLIDANTS
Examples of glidants include silicon dioxide, talc and corn-starch. A glidant is a substance that is added to a powder to improve its flowability. A glidant will only work at a certain range of concentrations. Above a certain concentration, the glidant will in fact function to inhibit flowability (which means that there's a critical concentration to be used if increasing powder's flowability is intended with respect to the glidant and the powder properties). In tablet manufacture, glidants are usually added just prior to compression [18].
Conventional Technique Used in the Preparation of Immediate Release Tablets:
- Tablet molding technique
- Direct compression technique
- Wet granulation technique
- Mass extrusion technique
- By solid dispersions
Tablet Molding
In this technology, water-soluble ingredients are used to disperse the tablet and disperse more quickly. The powder mixture is stirred with a hydro alcoholic solvent and molded into a tablet using a lower pressure than that used for conventional pill compression. The solvent was then released by air suspension. The molded tablets have a porous structure that promotes dissolution. Two common problems are mechanical power and the characteristics of hiding bad taste. Using binding agents such as sucrose, acacia or poly vinyl pyrrolidone may increase tablet performance. To counteract the anti-bad taste factor Van Scoik combined a compound containing various particles, which were sprayed with a concentrated mixture of molten hydrogenated cottonseed oil, sodium bicarbonate, lecithin, polyethylene glycol and an active tropical anticancer.
Direct Compression Method
In this way, the pills are pressed directly into the drug mixture and utensils without first treatment. The compressed mixture should have adequate flow areas and compact under pressure thus making pre-treatment such as wet acne unnecessary. Few drugs can be directly suppressed into acceptable quality pills. The type of disintegrant and its component are very important. Other factors to consider are the particle size distribution, the contact angle, the pore size distribution, the hardness of the tablet and the water absorption capacity. All of these factors determine the dispersion. Disintegrant augmentation technology is expensive and easy to use at the industrial level.
Wet Granulation Method
Wet granulation is the process of using a liquid binder to slightly compact the powder mixture. The amount of fluid should be controlled properly, as excessive wetting will cause the granules to become very hard and a little urination will make them more-soft and elastic. Aqueous solutions have the advantage of being safer to deal with than solvent-based systems but may not be suitable for drugs that are degraded by hydrolysis.
PROCEDURE
- The active ingredient and auxiliary ingredients are measured and mixed.
- Wet granulate is prepared by adding a liquid binder-adhesive to the powder mixture and mixing well. Examples of binding / adherence include liquid correction of cornstarch, natural gum-like gums, and cellulose extracts such as methyl cellulose, gelatin, and povidone.
- Check the humidity weight with matches to make pellets or granules.
- Drying granules. A regular tray-dryer or liquid bed dryer is widely used
- After the granules are dried, they are transferred to a screen smaller than the one used for wet weight to create granules of the same size.
Low-density wet shear processes use a very simple mixing tool, and can take a long time to achieve a uniform blend. Wet granulation shear processes use materials that mix powder and liquid at a rapid rate, and thus speed up the production process. Liquid bedding is a multi-step wetting process performed in the same vessel to pre-heat, melt, and dries the powders. It is used because it allows for close control of the granulation process.
Mass-Extrusion (Mass-Extrusion)
This technology involves softening the active blend using the solvent mixture of water-soluble polyethylene glycol and methanol and subsequent expulsion of softened mass through the extruder or syringe to get a cylinder of the product into even segments using heated blade to form tablets. The dried cylinder can also be used to coat granules for bitter drugs and thereby achieve taste masking.
By Solid Dispersions
When forming such a strong amorphous dispersion in solid dosage forms that are immediately released for oral administration such as the GI tract of a human-like animal, it is often desirable to increase the amount of dispersion present in the dosage form. This reduces the size of the solid volume form required to achieve the required volume. Depending on the dosage of the drug, it is generally desirable that the strong amorphous dispersion consist of at least 30 wt. %, preferably at least wt. %, and preferably at least 50 wt% or more in solid form. Such high-dose drug dispersal in the form of strong doses reduces the size of the dose form, makes it easier for the patient to swallow and often improves patient compliance.
Immediate dosage forms contain a strong dispersion that enhances the solubility of a “less soluble drug,” meaning that the drug may be “less soluble in water,” meaning that the drug has less water solubility at a physically fit pH. (e.g., pH 1-8) below 0.01 mg / mL, “slightly soluble in water,” that is, has a water solubility of up to 1 to 2 mg / mL, or even low to moderate water solubility, Melting in water from about 1 mg / mL up to about 20 to 40 mg / ml.
The dispersed drugs used to create the types of fast-release formulated loads include a solid distribution of the drug and one polymer to improve concentration at least. The polymer that promotes concentration is present in the dispersion used in the present in sufficient quantity to improve the saturation of the drug in the area of use associated with the formation of control.
At the very least, the dispersions used in the current establishment provide improved control-related focus that includes only glossy drugs. Therefore, the concentration-enhancing polymer is present in sufficient quantity so that when the dispersion is controlled in the applied area, the dispersion provides improved drug-related coagulation that incorporates an equal amount of crystalline drug, but without improving concentration of existing polymer [7].
EVALUATION OF IMMEDIATE RELEASE TABLETS
The blend is evaluated by following tests [19-21]
1. Angle of repose
2. Bulk density
3. Tapped density
4. Carr’s index
5. Hausner’s Ratio
1. Angle of Repose
Angle of repose was determined by using funnel method. The accurately weighed blend was taken in a funnel. The height of the funnel was adjusted in such a way that the tip of the funnel just touches the apex of the heap of blend. The drug excipient blend was allowed to flow through the funnel freely on to the surface. The diameter of the powder cone was measured and angle of repose was calculated using the following equation.
Tan = h/r
Where, h and r are the height and radius of the powder conc.
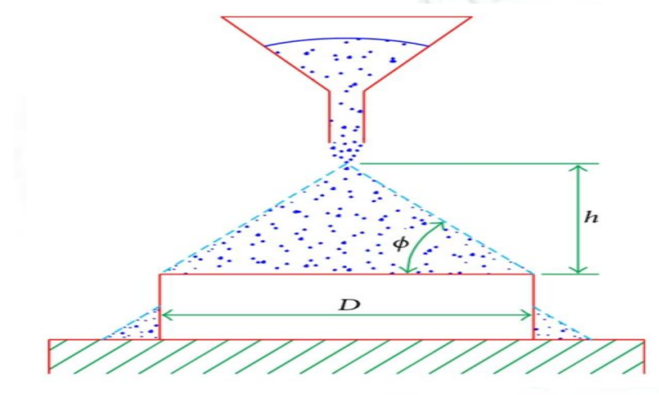
Figure 1: Measurement of angle of repose (fixed funnel method).
2. Bulk Density
Bulk density was determined by pouring a weighed quantity of tablet blend into graduated cylinder and measuring the height. Bulk density is the ratio of mass of tablet blend to bulk volume. It was calculated in gm/cm3 by the formula.
Bulk Density (BD)=Weight of granules (m) /untapped volume of granules (v).
Here; m=weight of powder or granules (gm)
v = Bulk Volume (cm.3)
3. Tapped Density
Tapped density is ratio of mass of tablet blend to tapped volume of tablet blend. Accurately weighed amount of tablet blend poured in graduated cylinder and height is measured. Then cylinder was allowed to 100tap under its own weight onto a hard surface. The tapping was continued until no further change in height was noted it was calculated in gm/cm3 by the formula.
Tapped Density (TD)=Weight of granules (m) /tapped volume of granules (v).
Here; m=weight of powder or granules (gm)
v=Tapped Volume (cm.3)
4. Compressibility Index
The Compressibility Index of the blends was determined by Carr’s compressibility index.
Carr’s compressibility index (%) = Compressibility is the ability of powder to decrease in volume under pressure using bulk density and tapped density the percentage compressibility of powder was determined, which is given as carr’s compressibility index.
It is indirectly related to the relative flow rate. Carr’s compressibility index was determined by the given formula.
Carr’s Index (%) = [(TBD-LBD) X 100] / TBD
4. Hauser’s ratio
Hausner’s ratio indicates the flow properties of powder and measured by the ratio of tapped density to bulk density. Hausner’s ratio was determined by the given Formula.
Hausner’s Ratio =Tapped density/ Poured density
Hausner’s ratio <1.25 - Good flow = Carr 1.25- Poor flow =33% Carr
In-vitro Evaluation of the prepared tablets
These tests are as following
- Appearance
- Thickness
- Hardness
- Weight variation
- Friability
- Disintegration
- Drug content
- In vitro Dissolution
- Stability studies
1. Appearance
The general appearance of tablet is its visual identity and all over elegance, shape, colour, surface textures. These all parameters are essential for consumer acceptance.
2. Thickness
The thickness of the tablets was determined by using vernier calipers. Randomly 10 tablets selected were used for determination of thickness that expressed in Mean± SD and unit is mm.
3. Hardness
The hardness of tablet is an indication of its strength against resistance of tablets to capping, abrasion or breakage under conditions of storage, transportation and handling before usage. Measuring the force required to break the tablet across tests it. Hardness of 10 tablets (randomly) from whole tablet batch was determined by Monsanto hardness tester. Hardness measured in kg/cm2.
4. Weight variation
The weight variation test is carried out in order to ensure uniformity in the weight of tablets in a batch. The total weight of 20 tablets randomly from whole batch was determined and the average was calculated. The individual weights of the tablets were also determined accurately and the weight variation was calculated.
5. Friability test
Friability is the loss of weight of tablet in the container due to removal of fine particles from the surface during transportation or handling. Roche friabilator was employed for finding the friability of the tablets. For tablets with an average weight of 0.65 g or less take a sample of whole tablets corresponding to about 6.5 g and for tablets with an average weight of more than 0.65 g take a sample of 10 whole tablets. Roche friabilator is rotated at 25rpm for 4 minutes for 100rounds. The tablets were dedusted and weighed again. The percentage of weight loss was calculated using the formula
%f= W0-W1/W0*100
%f = Percentage friability
W0 = Initial weight (Before test)
W1 = Final weight (After test)
6. Disintegration test
The USP device to rest disintegration was six glass tubes that are “3 long, open at the top, and held against 10” screen at the bottom end of the basket rack assembly. One tablet is placed in each tube and the basket rack is poisoned in 1 liter beaker of distilled water at 37± 2 o C, such that the tablets remain below the surface of the liquid on their upward movement and descend not closer than 2.5cm from the bottom of the beaker.
7. Drug content
10 tablets were powdered and 100mg drug equivalent powder dissolved in suitable media buffer or 0.1N HCl. Volume of the solution made up to 100ml by that media. Solution was filtered and diluted 100times and analyzed spectrophotometrically and further Calculation carried out to determine drug content in one tablet.
8. In vitro drug release studies
The immediate release tablets are subjected to in vitro drug release studies in pH 6.8 phosphate buffer or 0.1N HCl for 30 minutes to access the ability of the formulation for providing immediate drug delivery. Drug release studies were carried out in dissolution test apparatus using specified volume 900ml of dissolution media maintained at 37±100C. The tablets are kept in the cylindrical basket or directly placed in medium with paddle then rotated at 100 rpm. 5ml of the sample from the dissolution medium are withdrawn at each time interval (5, 10, 15 & 30 minutes) and 5ml of fresh medium was replaced each time. The samples were filtered and from the filtrate 1ml was taken and diluted to 10ml. These samples were analyzed spectrophotometrically and further calculation was carried out to get drug release.
RELEASE MODELS
Zero order release kinetics
It refers to the process of constant drug release from a drug delivery device independent of the concentration. In its simplest form, zero order release can be represented as
Q = Q0 + K0 t
Where Q is the amount of drug released or dissolved, Q0 is the initial amount of drug in solution (it is usually zero), and K0 is the zero-order release constant. The plot made: cumulative drug release vs. time. Graphical representation of fraction of drug dissolved verses time will be linear. The slope of the curve gives the value of K in zero order release kinetics. This is ideal behaviour for a dosage form and leads to minimum fluctuations in drug plasma levels. This is expressed mainly by osmotic pump systems and also transdermal systems, matrix tablets with low soluble drugs and coated forms [22].
First order release kinetics
The first order Equation describes the release from system where release rate is concentration dependent, expressed by the equation:
dc / dt = - Kt
Where K is first order rate constant expressed in units of time -1.
This equation can be expressed as:
Log Ct = Log C0 – k t / 2.303
Where, C0 is the initial concentration of drug and Ct is the concentration of drug in solution at time t. The equation predicts a first order dependence on the concentration gradient (C0 – Ct) between the static liquid layer next to the solid surface and the bulk liquid. The plot made: log cumulative of % drug remaining vs. time which would yield a straight line with a slope of –K/2.303. The dosage forms containing water soluble drug in porous matrices (Mulye and Turco, 1995) follows this profile such that the proportional to the amount of drug released by unit time diminishes.
Higuchi Model
The first example of a mathematical model aimed to describe drug release from a matrix system was proposed by Higuchi in 1963 this model is applicable to study the release of water soluble and low soluble drugs incorporated in semisolid and solid matrices.
Model expression is given by the equation:
Q = A [D (2C - Cs) Cs t] 1/2
Where Q is the amount of drug released in time t per unit area A, C is the drug initial concentration, Cs is the drug solubility in the media and D is the diffusivity of the drug molecules (diffusion coefficient) in the matrix [23].
Korsmeyer - Peppas Model
Korsmeyer et al (1983) derived a simple relationship which described drug release from a polymeric system. Ritger and Peppas and Korsmeyer and Peppas developed an empirical equation to analyse Fickian and non-Fickian release of drug from swelling as well as no swelling polymeric delivery systems.
To find out the mechanism of drug release, first 60% drug release data was fitted in Korsmeyer Peppas model
Mt / Mα = Ktn
Where Mt/Mα is fraction of drug released at time t, k is the rate constant (having units of tn) incorporating structural and geometric characteristics of the delivery system. n is the release exponent indicative of the mechanism of transport of drug through the polymer. The n value is used to characterize different release mechanisms.
CONCLUSION
A new dosage formula, an over-the-counter drug formula has been developed that offers integrated benefits for ease of administration and ease of dosing. These pills are designed to dispense medicines at an improved price. In order to meet these medical needs, the formulation has made significant efforts to develop forms of oral tablet therapy, which are dispersed and dissolved rapidly with improved dispersion. Immediate drug delivery does more benefits than standard dosage forms such as improved performance, faster bioavailability in the action set, better patient compliance. Expanding market diversification, which can be provided by issuing a quick dose form leads to increased revenue, while also targeting the number of patents that are poorly maintained and poorly managed. Due to the limitations of current technology, there is a need for advanced production processes for the rapid release of a robust form of medicine, which allows for easy handling and packaging and production costs similar to regular pills.
REFERENCES
- Notari R. (1980). Bio pharmaceutics and Clinical Pharmacokinetics, An Introduction, 3rd Ed. Marcel Dekker Inc. New York:52-54.
- Vinay K, Prajapati SK, Girish CS, Mahendra S, Neeraj k. (2012). Sustained release matrix type drug delivery system. World J Pharma Pharma Sci. 1(3):934-960.
- Kumar V, Sharma A, Sharma A, Joshi G, Dhillon V. (2011). Recent Advances in Novel Drug Delivery System for Delivery of Anti- Hypertensive Drugs. Int J D D Res. 3(1):252-259.
- Basu B, Bagadiya A, Makwana S, Vipul V, Batt D, Dharamsi A. (2011). Formulation and evaluation of fast dissolving tablets of cinnarizine using superdisintegrant blends and subliming material. J Adv Pharm Technol Res. 2(4):266-273.
- The Indian Pharmacopoeia. (2014). Government of India Ministry of health and family welfare, the Indian pharmacopoeia commission Ghaziabad:959.
- Sandeep N, Gupta MM. (2013). Fast Drug Release Dosage Form: A review. Journal of Drug Delivery & therapeutics. 3(2):155-161.
- Pahwa R, Gupta N. (2011). A Review: Super disintegrants in the development of orally disintegrating tablets. Int J Pharm Sci Res. 2(11):2767-2780.
- Satpute MM, Tour NS. (2013). Formulation and in vitro evaluation of fast dissolving tablets of metoprolol tartrate. Br J PharM Sci. 49(4).
- Arya A and Chandra A. (2010). Fast drug delivery system: A Review, Scholar Research Library. 2(2):350-361.
- Kilor V, Sapkal N, Awari J, Shewale B. (2010). Development And Characterization Of Enteric-Coated Immediate-Release Pellets Of Aceclofenac By Extrusion/Spheronization Technique Using Carrageenan As A Pelletizing Agent, AAPS Pharmscitech. 11(1):336-343.
- Aulton`S. (2001). Pharmaceutics, The Design & Manufacture Of Medicines, Biopharmaceutics And Pharmacokinetics, A Treatise, Second Edition, Valabh Prakashan:315-384.
- Syed A, et al. (2001). Immediate Release Drug Delivery Systems: A Review. Int J Biopharm Toxicol Res.
- Ansel`S. (2006). Pharmaceutical Dosage Forms & Drug Delivery Systems, Eighth Edition:227-260
- Sandeep N, Gupta MM. (2013). Immediate drug release dosage form: a review. J Drug Delivery & Therapeutics. 3(2):155-161.
- Rawlins EA. (2006). Tablets and Capsules. In: Bentleys. Text book of pharmaceutics. New Delhi: All India Traveller Publishers:234-310.
- Cooper J, Gunn C. (1986). Powder flow and compaction, In: Carter SJ, eds. Tutorial Pharmacy. CBS Publishers and Distributors, New Delhi, India: 211-233.
- Dedhiya et al. (2006). Lercanidipine immediate release composition. United States Patent Application.
- Chandira MR, Jayakar B, Pasupathi AC, Maruya PBL. (2009). Design, Development and Evaluation of Immediate Release Atorvastatin and Sustained Release Gliclazide Tablets. J Pharm Res. 6(2).
- Sood R, et al. (2012). Immediate release antihypertensive valsartan oral tablet: A Review. J Sci Res Pharm. 1(2):20-26.
- Pannala S, Rathnanand M. (2011). Preparation and in vitro evaluation of Nizatidine immediate release tablets. Int J Pharm Tech Res. 3(3):1688-1692.
- Dash S, Murthy PN, Nath L, Chowdhury P. (2010). Kinetic Modeling on drug release from controlled drug delivery systems. Acta Poloniae Pharmaceutica. 67(3):217-223.
- Higuchi T. (ᥣ). Mechanism of sustained action medication: Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 52:1145-1149.
- Singh J, Gupta S, Kaur H. (2011). Prediction of in vitro Drug Release Mechanisms form Extended Release Matrix Tablets using SSR/R2 Technique. Trends App Sci Res. 6(4):400-409.